Thomas J. Ebert
Phillip G. Schmid
Key Points
1. At equilibrium, the CNS partial pressure of inhaled anesthetics equals their arterial partial pressure, which in turn equals their alveolar partial pressure if cardiopulmonary function is normal.
2. Isoflurane is the most potent of the volatile anesthetics in clinical use, desflurane is the least soluble, and sevoflurane is the least irritating to the airways.
3. The inspired concentration and the blood:gas solubility of an inhaled anesthetic are the major determinants of the speed of induction. Solubility alone determines the rate of elimination, provided there is normal cardiopulmonary function.
4. Nitrous oxide can expand a pneumothorax to double or triple its size in 10 to 30 minutes, and washout of nitrous oxide can lower alveolar concentrations of oxygen and carbon dioxide, a phenomenon called diffusion hypoxia.
5. Minimum alveolar concentration (MAC) is the alveolar concentration of an inhaled anesthetic at one atmosphere that prevents movement in response to a surgical stimulus in 50% of patients. Concentrations of inhaled anesthetics that provide loss of awareness and recall are about 0.4 to 0.5 MAC.
6. Excluding data in patients <1 year of age (where MAC is lower than in older children), MAC decreases approximately 6% per decade.
7. Volatile anesthetics depress cerebral metabolic rate and increase cerebral blood flow (CBF) in a dose-dependent manner. The latter effect may increase intracranial pressure in patients with a mass-occupying lesion of the brain.
8. Hypocapnia may blunt or abolish volatile anesthetic-induced increases in CBF depending on when the hypocapnia is produced and the nature of the cerebral disease process.
9. Volatile anesthetics produce dose-dependent depression of the electroencephalogram, sensory-evoked potentials, and motor-evoked potentials.
10. Volatile anesthetics in current use decrease arterial blood pressure, systemic vascular resistance, and myocardial function comparably and in a dose-dependent fashion.
11. Volatile anesthetics decrease tidal volume, decrease ventilatory response to hypercarbia and hypoxia, increase respiratory rate, and relax airway smooth muscle in a dose-dependent fashion.
P.414
12. Unlike halothane, volatile anesthetics in current use have minimal adverse effects on the liver and might afford some protection for hepatocytes from ischemic and/or hypoxic injury.
13. Volatile anesthetics are potent triggers for malignant hyperthermia in genetically susceptible patients, while nitrous oxide is only a weak trigger.
14. CO2 absorbents degrade sevoflurane, desflurane, and isoflurane to carbon monoxide when the normal water content of the absorbent (13 to 15%) is markedly decreased (<5%).
Inhalation anesthetics are the most common drugs used for the provision of general anesthesia. Adding only a fraction of a volatile anesthetic to the inspired oxygen results in a state of unconsciousness and amnesia. When combined with intravenous adjuvants, such as opioids and benzodiazepines, a balanced technique is achieved that results in analgesia, further sedation/hypnosis, and amnesia. The popularity of the inhaled anesthetics for surgical procedures is because of their ease of administration and the ability to reliably monitor their effects with both clinical signs and end-tidal concentrations. In addition, the volatile anesthetic gases are relatively inexpensive in terms of the overall cost.
The most popular potent inhaled anesthetics used in adult surgical procedures are sevoflurane, desflurane, and isoflurane (Fig. 17-1). In pediatric cases, sevoflurane is most commonly employed. Although there are many similarities in terms of the overall effects of the volatile anesthetics (e.g., they all have a dose-dependent effect to decrease blood pressure), there are some unique differences that might influence the clinician's selection process depending on the patient's health and the surgical procedure. Discussion of the three most popular inhaled anesthetics provides the major emphasis of this chapter. For the sake of completeness and for historical perspective related to metabolism and toxicity, comments on halothane, enflurane, and methoxyflurane also are included.
|
|
|
Figure 17-1. Chemical structure of inhaled anesthetics. Halothane is an alkane, a halogen-substituted ethane derivative. It is no longer available commercially. Isoflurane and enflurane are isomers that are methyl ethyl ethers. Desflurane differs from isoflurane in the substitution of a fluorine for a chlorine atom and sevoflurane is a methyl isopropyl ether. |
Pharmacokinetic Principles
Kety1 in 1950 was the first to examine the pharmacokinetics of inhaled agents in a systematic fashion. Eger2 accomplished much of the early research in the field, leading to his landmark text on the subject in 1974. The inhaled anesthetics differ substantially from nearly all other drugs because they are gases given via inhalation. This makes their pharmacokinetics unique as well, and thus discussion of pharmacokinetic principles of currently used agents is necessary for understanding and predicting their effects.
Drug pharmacology is classically divided into two disciplines, pharmacodynamics and pharmacokinetics. Pharmacodynamics can be defined as what drugs do to the body. It describes the desired and undesired effects of drugs, as well as the cellular and molecular changes leading to these effects. Pharmacokinetics can be defined as what the body does to drugs. It describes where drugs go, how they are transformed, and the cellular and molecular mechanisms underlying these processes.
Tissues are often grouped into hypothetical compartments based on perfusion. An important implication of different compartments and perfusion rates is the concept of redistribution. After a given amount of drug is administered, it reaches highly perfused tissue compartments first, where it can equilibrate rapidly and exert its effects. With time, however, compartments with lower perfusion rates receive the drug and additional equilibria are established between blood and these tissues. As the tissues with lower perfusion absorb drug, maintenance of equilibria throughout the body requires drug transfer from highly perfused compartments back into the bloodstream. This lowering of drug concentration in one compartment by delivery into another compartment is called redistribution.
In discussions of the inhaled anesthetics, the absorption phase is usually called uptake, the metabolic phase is usually called biotransformation, and the excretion phase is usually called elimination.
Unique Features of Inhaled Anesthetics
Speed, Gas State, and Route of Administration
The inhaled anesthetics are among the most rapidly acting drugs in existence, and when administering a general anesthetic, this speed provides a margin of safety. The ability to quickly increase or decrease anesthetic levels as necessary can mean the difference between an anesthetic state and an anesthetic misadventure. Speed also means efficiency. Rapid induction and recovery may lead to faster operating room turnover times, shorter recovery room stays, and earlier discharges to home.
Technically, of the inhaled anesthetics only nitrous oxide and xenon are true gases, while the so-called potent agents are the vapors of volatile liquids. But for simplicity, all of them are referred to as gases because they are all in the gas phase when administered via the lungs. As gases, none deviate significantly from ideal gas behavior. These agents are all non-ionized and have low molecular weights. This allows them to diffuse rapidly without the need for facilitated diffusion or active
P.415
transport from bloodstream to tissues. The other advantage of gases is that they can be delivered to the bloodstream via a unique route available in all patients: the lungs.
|
Table 17-1 Physiochemical Properties of Volatile Anesthetics |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Speed, gaseous state, and the lung route of administration combine to form the major beneficial feature of the inhaled anesthetics: the ability to decrease plasma concentrations as easily and as rapidly as they are increased.
Physical Characteristics of Inhaled Anesthetics
The physical characteristics of inhaled anesthetics are shown in Table 17-1. The goal of delivering inhaled anesthetics is to produce the anesthetic state by establishing a specific concentration of anesthetic molecules in the central nervous system (CNS). This is done by establishing the specific partial pressure of the agent in the lungs, which ultimately equilibrates with the brain and spinal cord. At equilibrium, CNS partial pressure equals blood partial pressure, which in turn equals alveolar partial pressure:
![]()
where P is partial pressure. Equilibration is a result of three factors:
1. Inhaled anesthetics are gases rapidly transferred bidirectionally via the lungs to and from the bloodstream and subsequently to and from CNS tissues as partial pressures equilibrate.
2. Plasma and tissues have a low capacity to absorb the inhaled anesthetics relative to the amount we can deliver to the lungs, allowing us to quickly establish or abolish anesthetizing concentrations of anesthetic in the bloodstream and ultimately the CNS.
3. Metabolism, excretion, and redistribution of the inhaled anesthetics are minimal relative to the rate at which they are delivered or removed from the lungs. This permits easy maintenance of blood and CNS concentrations.
The so-called permanent gases, such as oxygen and nitrogen, exist only as gases at ambient temperatures. Gases such as nitrous oxide can be compressed into liquids under high pressure at ambient temperature. Most potent volatile anesthetics are liquids at ambient temperature and pressure. If the system in which the volatile liquid resides is a closed container, molecules of the substance will equilibrate between the liquid and gas phases. At equilibrium, the pressure exerted by molecular collisions of the gas against the container walls is the vapor pressure. One important property of vapor pressure is that as long as any liquid remains in the container, the vapor pressure is independent of the volume of that liquid. As with any gas, however, vapor pressure is proportional to temperature.
For all of the potent agents, at 20°C the vapor pressure is below atmospheric pressure. If the temperature is raised, the vapor pressure increases. The boiling point of a liquid is the temperature at which its vapor pressure exceeds atmospheric pressure in an open container. Desflurane is bottled in a special container because its boiling point of 23.5°C makes it boil at typical room temperatures. Boiling does not occur within the bottle because it is countered by buildup of vapor pressure within the bottle, but once opened to air, the desflurane would quickly boil away. The bottle is designed to allow transfer of desflurane from bottle to vaporizer without exposure to the atmosphere.
Gases in Mixtures
For any mixture of gases in a closed container, each gas exerts a pressure proportional to its fractional mass. This is its partial
P.416
pressure. The sum of the partial pressures of each gas in a mixture of gases equals the total pressure of the entire mixture (Dalton's law).
![]()
Another way to state this is that each gas in a mixture of gases at a given volume and temperature has a partial pressure that is the pressure it would have if it alone occupied the volume. The entire mixture behaves just as if it were a single gas according to the ideal gas law.
Gases in Solution
Partial pressure of a gas in solution is a bit complex because pressure can only be measured in the gas phase, while in solution the amount of gas is measured as a concentration. Partial pressure of a gas in solution refers to the pressure of the gas in the gas phase (if it were present) in equilibrium with the liquid. It is important to talk of partial pressures, however, because gases equilibrate based on partial pressures, not concentrations.
Gas molecules within a liquid interact with solvent molecules to a much larger extent than do molecules in the gas phase. Solubility is the term used to describe the tendency of a gas to equilibrate with a solution, hence determining its concentration in solution. Henry's law expresses the relationship of concentration of a gas in solution to the partial pressure of the gas with which the solution is in equilibrium:
![]()
where Cg is concentration of gas in solution, k is a solubility constant, and Pg is the partial pressure of the gas. From Equation 17-3 one can see that doubling the pressure of a gas doubles its concentration in solution. A more clinically useful expression of solubility is the solubility coefficient, λ:
![]()
where V = volume. This equation states that for any gas in equilibrium with a liquid, a certain volume of that gas dissolves in a given volume of liquid.
The principles of partial pressures and solubility apply in mixtures of gases in solution. That is, the concentration of any one gas in a mixture of gases in solution depends on two factors: (1) its partial pressure in the gas phase in equilibrium with the solution, and (2) its solubility within that solution.
The implications of these properties are that anesthetic gases administered via the lungs diffuse into blood until the partial pressures in alveoli and blood are equal. The concentration of anesthetic in the blood depends on the partial pressure at equilibrium and the blood solubility. Likewise, transfer of anesthetic from blood to target tissues also proceeds toward equalizing partial pressures, but at this interface there is no gas phase. A partial pressure still exists to force anesthetic molecules out of solution and into a gas phase but there is no gas phase because blood (outside the lungs) and tissues are like closed, liquid-filled containers. Remember the principle: the partial pressure of a gas in solution represents the pressure that the gas in equilibrium with the liquid would have if a gas phase existed in contact with the liquid phase.
The concentration of anesthetic in target tissue depends on the partial pressure at equilibrium and the target tissue solubility. Because inhaled anesthetics are gases, and because partial pressures of gases equilibrate throughout a system, monitoring the alveolar concentration of inhaled anesthetics provides an index of their effects in the brain.
In summary:
1. Inhaled anesthetics equilibrate based on their partial pressures in each tissue (or tissue compartment), not based on their concentrations.
2. The partial pressure of a gas in solution is defined by the partial pressure in the gas phase with which it is in equilibrium. Where there is no gas phase the partial pressure reflects a force to escape out of solution.
3. The concentration of anesthetic in a tissue depends on its partial pressure and tissue solubility.
Finally, the particular terminology used when referring to gases in the gas phase or absorbed in plasma or tissues is important. Inspired concentrations or fractional volumes of inhaled anesthetic are typically used rather than partial pressure. Partial pressure is expressed in millimeters of mercury (mm Hg) or torr (1 torr = 1 mm Hg) or kilopascals (kPa). For most drugs, concentration is expressed as mass (milligram [mg]) per volume (milliliter [mL]), but it can also be expressed in percent by weight or volume. Because volume of a gas in the gas phase is directly proportional to mass according to the ideal gas law, it is easier to express this fractional concentration as a percent by volume. In the gas phase, fractional concentration is equal to the partial pressure divided by ambient pressure, usually atmospheric, or:
![]()
Anesthetic Transfer: Machine to Central Nervous System
When the fresh gas flow and the vaporizer are turned on, fresh gas with a fixed fractional concentration of anesthetic leaves the fresh gas outlet and mixes with the gas in the circuit—the bag, tubing, absorbent canister, and piping. It is immediately diluted to a lower fractional concentration, then slowly rises as this compartment equilibrates with the fresh gas flow. With spontaneous patient ventilation by mask, the anesthetic gas passes from circuit to airways. The fractional concentration of anesthetic leaving the circuit is designated as FI(fraction inspired). In the lungs the gas comprising the dead space in the airways (trachea, bronchi) and the alveoli further dilutes the circuit gas. The fractional concentration of anesthetic present in the alveoli is FA (fraction alveolar). The anesthetic then passes across the alveolar–capillary membrane and dissolves in pulmonary blood according to the partial pressure of the gas and its blood solubility. It is further diluted and travels via bulk blood flow throughout the vascular tree. The anesthetic then passes via simple diffusion from blood to tissues as well as between tissues.
The vascular system delivers blood to three physiologic tissue groups; the vessel-rich group (VRG), the muscle group, and the fat group. The VRG includes the brain, heart, kidney, liver, digestive tract, and glandular tissues. The percent of body mass and perfusion of each group are shown in Table 17-2. The CNS tissues of the VRG are referred to as tissues of desired effect. The other tissues of the VRG that comprise the compartment are referred to as tissues of undesired effects. The tissues of the muscle and fat groups comprise thetissues of accumulation.
Anesthetic is delivered most rapidly to the VRG because of high blood flow. Here it diffuses according to partial pressure gradients. CNS tissue takes in the anesthetic according to the tissue solubility, and at a high enough tissue concentration, unconsciousness and anesthesia are achieved. Increasing CNS tissue concentrations cause progressively deeper stages of anesthesia. As this is occurring, anesthetic is also distributing to other VRG tissues. Also coincident with delivery to the
P.417
CNS, anesthetic is being delivered—albeit more slowly because of lower perfusion—to muscle and fat, where it accumulates and may affect the speed of emergence from the anesthetic. In reality, the fat solubilities provide little influence on emergence in cases lasting <4 hours since the delivery of anesthetic to fat tissue is extremely slow as a result of low blood flow. The concentration of inhaled anesthetic in a given tissue at a particular time during the administration depends not only on tissue blood flow, but also on tissue solubility, which governs how the inhaled anesthetics partition themselves between blood and tissue. Partitioning depends on the relative solubilities of the anesthetic for each compartment. These relative solubilities are expressed by a partition coefficient, δ, which is the ratio of dissolved gas (by volume) in two-tissue compartments at equilibrium. Some of the partition coefficients for the inhaled anesthetics are shown in Table 17-1.
|
Table 17-2 Distribution of Cardiac Output by Tissue Group |
||||||||||||||||
|
Uptake and Distribution
FA/FI
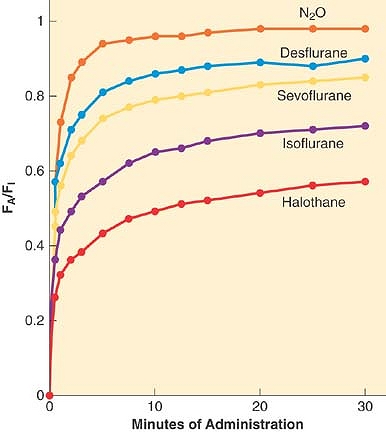
A simple, common way to assess anesthetic uptake is to follow the ratio of fractional concentration of alveolar anesthetic to inspired anesthetic (FA/FI) over time. Experimentally derived data for FA/FI versus time during induction are shown in Figure 17-2. The faster FA rises relative to FI, the faster the speed of induction since FA is proportional to PA (FA =PA/Pbarometric) and PA = Pblood = PCNS; that is, the alveolar fraction is directly proportional to the partial pressure of anesthetic in the CNS.
As fresh gas carrying anesthetic begins to flow into the air-filled circuit (assuming complete mixing), the concentration in the circuit (FI) will rise according to first-order kinetics:
![]()
FFGO is the fraction of inspired anesthetic in the gas leaving the fresh gas outlet (i.e., the vaporizer setting), T is time, and τ is a time constant. The time constant is simply the volume or “capacity” of the circuit (VC) divided by the fresh gas flow (FGF) or τ = VC/FGF. For example, if the bag, tubing, absorbent canister, and piping comprise 8 L, and the fresh gas flow is 2 L, the time constant τ = 8/2 = 4. One of the characteristics of first-order kinetics is that 95% of maximum is reached after three time constants—in this case, 3 × 4 = 12 minutes.
Because 12 minutes is relatively long, starting with a higher FFGO can increase the rate of rise of FI. Using the earlier example with τ = 4, by first-order kinetics 63% of maximum is reached after one time constant, or 4 minutes. To attain an FI of 2% at 4 instead of 12 minutes, the FFGO can be set to 3.2% (2% divided by 0.63) and then lowered to 2% at the 4-minute mark.
Other ways to speed the increase in FI include increasing the fresh gas flow, thus decreasing τ. Furthermore, the rebreathing bag can be collapsed prior to starting the fresh gas flow, such that the capacity in the circuit (VC) is less, which also decreases τ. Finally, at high flows (>4 L/min) there is far less mixing because fresh gas pushes “old” gas out of the circuit via the pop-off valve before complete mixing occurs, causing FI to increase at a greater rate; this is the most important factor in rapidly increasing FI to the desired concentration.
|
|
|
Figure 17-2. The rise in alveolar (FA) anesthetic concentration toward the inspired (FI) concentration is most rapid with the least soluble anesthetics, nitrous oxide (N2O), desflurane, and sevoflurane. It rises most slowly with the more soluble anesthetics, such as halothane. All data are from human studies. (Adapted from Yasuda N, Lockhart SH, Eger EI II, et al: Comparison of kinetics of sevoflurane and isoflurane in humans. Anesth Analg 1991; 72: 316; and Yasuda N, Lockhart SH, Eger EI II, et al: Kinetics of desflurane, isoflurane, and halothane in humans. Anesthesiology 1991; 74: 489.) |
One factor that delays the rate of rise of FI is that CO2 absorbent can adsorb and decompose the inhaled anesthetics. From a practical standpoint, this does not affect the rate of rise in FI to a significant extent compared with other factors. Another factor that delays the rate of rise of FI is solubility of the inhaled anesthetics in some of the plastic and rubber parts of the anesthesia circuit. This absorption has been quantified, but plays only a small role in decreasing the rate of rise of FI.
Rise in FA in the Absence of Uptake
The rate of rise in FI discussed earlier assumes that no anesthetic is mixing with gas in the patient's lungs. In reality, circuit gas mixes with exhaled gases from the lung with each breath, thus lowering FI within the circuit. If high fresh gas flows (>4 L/min), which produce a high volume of gas at the desired concentration, are used, little mixing with exhaled air occurs and FI is relatively fixed. In this situation, circuit gas enters the lungs where it mixes with alveolar gas. If there were no blood flow to the lungs, FA would rise in a fashion analogous to FI; that is:
![]()
In this equation, τ is the time constant for alveolar rise in anesthetic concentration and equals the functional residual capacity (FRC) of the patient's lungs divided by minute ventilation, [V with dot above]A. There are two ways to speed the equilibration of FA with FI, that is, to decrease τ. One way is to increase minute ventilation,
P.418
and the other is to decrease FRC. Both of these methods can be used to speed induction by mask: the patient can exhale deeply before applying the mask (to decrease the initial FRC), and the patient can breathe deeply and rapidly (to increase) after the mask is applied. Importantly, high alveolar ventilation relative to uptake from the lungs to the bloodstream generates the initial high slope to the curves shown in Figure 17-2.
|
Table 17-3 Factors that Increase or Decrease the Rate of Rise of FA/FI |
||||||||||||||||||
|
||||||||||||||||||
One of the reasons that pediatric inductions by spontaneous breathing of inhaled anesthetics are so much quicker than adult inductions is that the low FRC relative to [V with dot above]A of children makes for a low time constant, and hence a more rapid increase in FA/FI. One important caveat about the relationship of FA to FRC is that FRC includes airway dead space; thus, in reality, FA by Equation 17-7 is not just the concentration of inhaled anesthetic in the alveoli but also the concentration in the entire lung. However, it is simply called the alveolar concentration because the dead space in the airways is relatively insignificant and only the alveolar gas is exchanging anesthetic with the blood.
Rise in FA in the Presence of Uptake
Anesthetics are soluble in tissues, thus uptake of anesthetic from alveoli to blood is again characterized by first-order kinetics:
![]()
Here, PB is the barometric pressure and the time constant, τ, equals “capacity” (volume of anesthetic dissolved in blood at the desired alveolar partial pressure) divided by flow (volume of anesthetic delivered per unit time). For any given flow of anesthetic into the system, this capacity for the more soluble halothane is greater than the capacity for the less soluble desflurane; thus, τ for halothane is greater than that for desflurane. The more soluble the inhaled anesthetic, the larger the capacity of the blood and tissues for that anesthetic, and the longer it takes to saturate at any given delivery rate.
The most important factor in the rate of rise of FA/FI is uptake of anesthetic from the alveoli into the bloodstream. The rate of rise of FA/FI (especially the position of the “knees” in the curves of Figure 17-2) reflects the speed at which alveolar anesthetic (FA) equilibrates with that being delivered to the lungs (FI). Since there is uptake from alveoli to blood, FA is not solely a function of FI and time. The greater the uptake, the slower the rate of rise of FA/FI, and vice versa. Since uptake is proportional to tissue solubility, the less soluble the anesthetic (such as desflurane), the lesser its uptake and the faster it reaches equilibrium, PA = Pblood = PCNS.
Consider a hypothetical example. Suppose that halothane and desflurane are soluble in blood, but insoluble in all other tissues. Suppose further that total lung capacity and blood volume were both 5 L. If a fixed volume of anesthetic is delivered to the lungs (by asking the patient to take one deep breath and hold it), according to the blood:gas partition coefficients for halothane (2.5) and desflurane (0.42), 71.4% of the delivered halothane will be transferred to the blood while 28.6% remains in the alveoli (71.4/28.6 = 2.5). In contrast, 29.6% of the desflurane will be transferred to the blood while 70.4% remains in the alveoli (29.6/70.4 = 0.42). Therefore, 2.4 times (71.4/29.6) more halothane than desflurane (by volume or number of molecules) will be transferred from alveoli to bloodstream before partial pressures equilibrate. At equilibrium, the alveolar partial pressures of halothane and desflurane are 28.6% and 70.4% of their inhaled values, respectively. This means that FA rises faster with desflurane than halothane, as does FA/FI.
Blood uptake of anesthetic is expressed by the equation:
![]()
where [V with dot above]B is blood uptake, δb/g is the blood:gas partition coefficient, Q is cardiac output, PA is alveolar partial pressure of anesthetic, Pv is mixed venous partial pressure of anesthetic, and PB is barometric pressure. This is the Fick equation applied to blood uptake of inhaled anesthetics. The greater the value of V.B, the greater the uptake from alveoli to blood, and the slower the rise in FA/FI.
From the preceding paragraphs, the parameters that increase or decrease the rate of rise in FA/FI during induction can now be clearly delineated and these important factors have been substantiated in experimental models (Table 17-3).
Distribution (Tissue Uptake)
The maximum FA/FI at a given inspired concentration of anesthetic, cardiac output, and minute ventilation depends entirely on the solubility of that drug in the blood as characterized by the blood:gas partition coefficient δb/g. This can be seen in the time curves for the rise in FA/FI during induction for the various inhalation anesthetics shown inFigure 17-2. The first “knee” in each curve in Figure 17-2 represents the point at which the rapid rise in Pv begins to taper off; that is, when significant inhaled anesthetic concentrations begin to build up in the bloodstream because of distribution to and equilibration with the various tissue compartments.
P.419
As blood is equilibrating with alveolar gas, it also begins to equilibrate with the VRG, muscle, and, more gradually, the fat compartments based on perfusion. Muscle is not that different from the VRG, having partition coefficients that range from 1.2 (nitrous oxide) to 3.4 (halothane), just under a threefold difference; and for each anesthetic except nitrous oxide, the muscle partition coefficient is approximately double that for the VRG. Although both VRG and muscle are lean tissues, the muscle compartment equilibrates far more slowly than the VRG. The explanation comes in part due to the mass of the compartments relative to perfusion. The perfusion of the VRG is about 75 mL/min/100 g of tissue, whereas it is only 3 mL/min/100 g of tissue in the muscle (Table 17-2). This 25-fold difference in perfusion between VRG (especially brain) and muscle means that even if the partition coefficients were equal, the muscle would still take 25 times longer to equilibrate with blood.
Fat is perfused to a lesser extent than muscle and its time for equilibration with blood is considerably slower because the partition coefficients are so much greater. All of the potent agents are highly lipid-soluble. Partition coefficients range from 27 (desflurane) to 51 (halothane). On average, the solubility for these agents is about 25 times greater in fat than in the VRG group. Thus, fat equilibrates far more slowly with the blood and does not play a significant role in determining speed of induction. After long anesthetic exposures (>4 hours), the high saturation of fat tissue may play a role in delaying emergence.
Nitrous oxide represents an exception. Its partition coefficients are fairly similar in each tissue: it does not accumulate to any great extent and is not a very potent anesthetic. Its utility lies as an adjunct to the potent agents, and as a vehicle to speed induction.
Metabolism
Data suggest that enzymes responsible for biotransformation of inhaled anesthetics become saturated at less than anesthetizing doses of these drugs, such that metabolism plays little role in opposing induction. It may, however, have some significance to recovery from anesthesia, as discussed later.
Overpressurization and the Concentration Effect
There are several ways to speed uptake and induction of anesthesia with the inhaled anesthetics. The first is overpressurization, which is analogous to an intravenous bolus. This is the administration of a higher partial pressure of anesthetic than the alveolar concentration (FA) actually desired for the patient. Inspired anesthetic concentration (FI) can influence both FA and the rate of rise of FA/FI. The greater the inspired concentration of an inhaled anesthetic, the greater the rate of rise. This concentration effect has two components: the concentrating effect and an augmented gas inflow effect.
For example, consider the administration of 10% anesthetic (10 parts anesthetic and 90 parts other gas) to a patient in whom 50% of the anesthetic in the alveoli is absorbed by the blood. In this case, five parts (0.5 × 10) anesthetic remain in the alveoli, five parts enter the blood, and 90 parts remain as other alveolar gas. The alveolar concentration is now 5/(90 + 5) = 5.3%. Consider next administering 50% anesthetic with the same 50% uptake. Now 25 parts anesthetic remain in alveoli, 25 parts pass into blood, and 50 parts remain as other alveolar gas. The alveolar concentration becomes 25/(50 + 25) = 33%. Giving 5 times as much anesthetic has led to a 33%/5.3% = 6.2 times greater alveolar concentration. The higher the FI, the greater the effect. Thus nitrous oxide, typically given in concentrations of 50 to 70%, has the greatest concentrating effect. This is why the FA/FI versus time curve inFigure 17-2 rises the most quickly with nitrous oxide, even though desflurane has a slightly lower blood:gas solubility.
This is not the complete picture; there is yet another factor to consider. As gas is leaving the alveoli for the blood, new gas at the original FI is entering the lungs to replace that which is taken up by the blood. This other aspect of the concentration effect has been called augmented gas inflow. Again, take the example of 10% anesthetic delivered with 50% uptake into the bloodstream. The five parts anesthetic absorbed by the bloodstream are replaced by gas in the circuit that is still 10% anesthetic. The five parts anesthetic and 90 parts other gas left in the lungs mix with five parts replacement gas, or 5 × 0.10 = 0.5 parts anesthetic. Now the alveolar concentration is (5 + 0.5)/(100) = 5.5% (as compared to 5.3% without augmented inflow). For 50% anesthetic and 50% uptake, 25 parts of anesthetic removed from the alveoli are replaced with 25 parts of 50% anesthetic, giving a new alveolar concentration of (25 + 12.5)/(100) = 37.5% (as compared to 33% without augmented inflow). Thus, 5 times the FI leads to 37.5/5.5 = 6.8 times greater FA (compared to 6.2 times without augmented gas inflow). Of course, this cycle of absorbed gas being replaced by fresh gas inflow is continuous and has a finite rate, so our example is a simplification.
Second Gas Effect
A special case of concentration effect applies to administration of a potent anesthetic with nitrous oxide—that is, two gases simultaneously. Along with the concentration of potent agent in the alveoli via its uptake, there is further concentration via the uptake of nitrous oxide, a process called the second gas effect. The principle is simple (Figs. 17-3 and 17-4). Consider,
P.420
for example, administering 2% of a potent anesthetic in 70% nitrous oxide and 28% oxygen. In this case, nitrous oxide, with its extremely high partial pressure (despite low solubility), partitions into the blood more rapidly than the potent anesthetic, decreasing the alveolar N2O (nitrous oxide) concentration by some amount (e.g., by 50%). Ignoring uptake of the potent anesthetic, the uptake of N2O is 35 parts, leaving 35 parts N2O, 28 parts O2, and two parts potent agent in the alveoli. The anesthetic gas is now present in the alveoli at a concentration of 2/(2 + 35 + 28) = 3.1%. The potent agent has been concentrated and FA is increased.
|
|
|
Figure 17-3. The concentration effect is demonstrated in the top half of the graph from dogs receiving nitrous oxide (N2O). Administration of 70% nitrous oxide produces a more rapid rise in the FA/FI ratio of nitrous oxide than administration of 10% nitrous oxide. The second gas effect is demonstrated in the lower graphs. The FA/FI ratio for 0.5% halothane rises more rapidly when given with 70% nitrous oxide than when given with 10% nitrous oxide. (Adapted from Epstein R, Rackow H, Salanitre E, et al: Influence of the concentration effect on the uptake of anesthetic mixtures: The second gas effect. Anesthesiology 1964; 25: 364.) |
|
|
|
Figure 17-4. A graphic and relative equation to demonstrate the second-gas effect. In this hypothetical example, the second gas is set at 2% of a potent anesthetic and the model is set for 50% uptake of the first gas (nitrous oxide [N2O]) in the first inspired breath. The second gas is concentrated because of the uptake of N2O (middle panel). On replenishing the inspired second gas (FI = 2%) in the next breath, the second gas has been concentrated to be 2.7% because of the uptake of N2O in the previous breath. |
Ventilation Effects
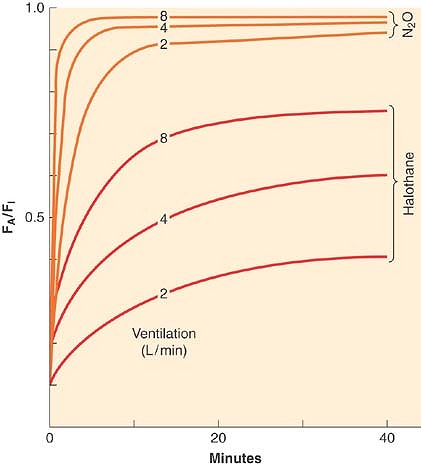
As indicated by Figure 17-2 and Table 17-3, inhaled anesthetics with very low tissue solubility have an extremely rapid rise in FA/FI with induction. This suggests that there is very little room to improve this rate by increasing or decreasing ventilation, which is consistent with the experimental evidence shown in Figure 17-5. The greater the solubility of an inhaled anesthetic, the more rapidly it is absorbed by the bloodstream, such that anesthetic delivery to the lungs may be rate limiting to the rise in FA/FI. Therefore, for more soluble anesthetics, augmentation of anesthetic delivery by increasing minute ventilation also increases the rate of rise in FA/FI.
Spontaneous minute ventilation is not static, however, and to the extent that the inhaled anesthetics depress spontaneous ventilation with increasing inspired concentration, [V with dot above]A will decrease and so will the rate of rise of FA/FI. This is demonstrated in Figure 17-5. This negative feedback should not be considered a drawback of the inhaled anesthetics because the respiratory depression produced at high anesthetic concentrations essentially slows the rise in FA/FI. This might arguably add a margin of safety in preventing an overdose. Controlled ventilation does not offer this margin of safety.
Perfusion Effects
As with ventilation, cardiac output is not static during the course of induction. For the less soluble agents, changes in cardiac output do not affect the rate of rise of FA/FI to a great extent, but for the more soluble agents the effect is noticeable, as seen in Figure 17-6. However, as inspired concentration increases, greater cardiovascular depression reduces anesthetic uptake and actually increases the rate of rise of FA/FI. This positive feedback can rapidly lead to profound cardiovascular depression. Figure 17-6 presents experimental data in which lower cardiac outputs lead to a much more rapid rise in FA/FI when [V with dot above]A is held constant. This more rapid rise is greater than can be accounted for just by concentration effect.
|
|
|
Figure 17-5. The FA/FI ratio rises more rapidly if ventilation is increased from 2 to 8 L/min. Solubility modifies this impact of ventilation; for example, the effect is greatest with the least-soluble anesthetic, nitrous oxide (N2O; top three lines), and least with the more soluble anesthetic, halothane. (Adapted from Eger EI II: Ventilation, circulation and uptake, Anesthetic Uptake and Action. Baltimore, Williams & Wilkins, 1974, pp 122.) |
P.421
|
|
|
Figure 17-6. If ventilation is fixed, an increase in cardiac output from 2 to 18 L/min will decrease the alveolar anesthetic concentration by augmenting uptake, thereby slowing the rise of the FA/FI ratio. This effect is most prominent with the more soluble anesthetics (halothane) than with the less soluble anesthetics (nitrous oxide [N2O]). (Adapted from Eger EI II: Ventilation, circulation and uptake, Anesthetic Uptake and Action. Baltimore, Williams & Wilkins, 1974, p 131.) |
Ventilation–Perfusion Mismatching
Ventilation and perfusion are normally fairly well matched in healthy patients such that PA (alveolar partial pressure)/PI and Pa (arterial partial pressure)/PI are the same curve. However, if significant intrapulmonary shunt occurs, as in the case of inadvertent bronchial intubation, the rate of rise of alveolar and arterial anesthetic partial pressures can be affected. The effects, however, depend on the solubility of the anesthetic, as seen in Figure 17-7. Ventilation of the intubated lung is dramatically increased while perfusion increases slightly. The nonintubated lung receives no ventilation, while perfusion decreases slightly. For the less-soluble anesthetics, increased ventilation of the intubated lung cannot appreciably increase alveolar partial pressure relative to inspired concentration on that side, but alveolar partial pressure on the nonintubated side is essentially zero. Pulmonary mixed venous blood, therefore, comprises nearly equal parts blood containing normal amounts of anesthetic and blood containing no anesthetic; that is, diluted relative to normal. Thus the rate of rise in Pa relative to PI is significantly reduced. There is less total anesthetic uptake, so the rate of rise of PA relative to PI increases even though induction of anesthesia is slowed because CNS partial pressure equilibrates with Pa. For the more soluble anesthetics, increased ventilation of the intubated lung does increase the alveolar partial pressure relative to inspired concentration on that side. Pulmonary venous blood from the intubated side contains a higher concentration of anesthetic that lessens the dilution by blood from the nonintubated side. Thus the rate of rise of Pa/PI is not as depressed as that for the less soluble anesthetics, and induction of anesthesia is less delayed relative to normal.
Elimination
Percutaneous and Visceral Loss
Although the loss of inhaled anesthetics via the skin is very small, it does occur and the loss is the greatest for nitrous oxide. These anesthetics also pass across gastrointestinal viscera and the pleura. During open abdominal or thoracic surgery there is some anesthetic loss via these routes. Relative to losses by all other routes, losses via percutaneous and visceral routes are insignificant.
Diffusion Between Tissues
Using more elaborate mathematical modeling of inhaled anesthetic pharmacokinetics than presented here, several laboratories have derived a five-compartment model that best describes tissue compartments. These compartments are the alveoli, the VRG, the muscle, the fat, and one additional compartment. Current opinion is that this fifth compartment represents adipose tissue adjacent to lean tissue that receives anesthetic via intertissue diffusion. This transfer of anesthetic is not insignificant, and may account for up to one third of uptake during long administration.
Exhalation and Recovery
Recovery from anesthesia, like induction, depends on anesthetic solubility, cardiac output, and minute ventilation. Solubility is the primary determinant of the rate of fall of FA
P.422
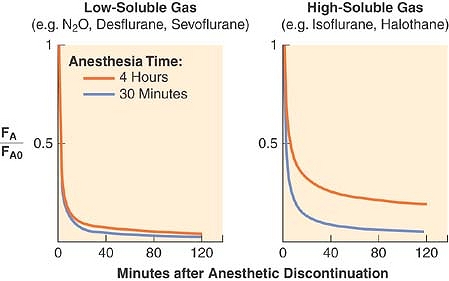
(Fig. 17-8). The greater the solubility of inhaled anesthetic, the larger the capacity for absorption in the bloodstream and tissues. The “reservoir” of anesthetic in the body at the end of administration depends on tissue solubility (which determines the capacity) and the dose and duration of anesthetic (which determine how much of that capacity is filled). Recovery from anesthesia, or “washout,” is usually expressed as the ratio of expired fractional concentration of anesthetic (FA) to the expired concentration at time zero (FA0) when the anesthetic was discontinued (or FA/FA0). Elimination curves of low- and high-soluble anesthetics are shown in Figure 17-9. The longer the duration of a highly soluble anesthetic, the greater the reservoir of anesthetic in the body, and the higher the curve seen in the right half of Figure 17-9. This effect is nearly absent with low-soluble agents such as nitrous oxide, desflurane, and sevoflurane.3
|
|
|
Figure 17-7. When no ventilation/perfusion abnormalities exist, the alveolar (PA) or end-tidal (PET) and arterial (Pa) anesthetic partial pressures rise together (blue lines) toward the inspired partial pressure (PI). When 50% of the cardiac output is shunted through the lungs, the rate of rise of the end-tidal partial pressure (orange lines) is accelerated while the rate of rise of the arterial partial pressure (green lines) is slowed. The greatest effect of shunting is found with the least soluble anesthetics. (Adapted from Eger EI II, Severinghaus JW: Effect of uneven pulmonary distribution of blood and gas on induction with inhalation anesthetics. Anesthesiology 1964; 25: 620.) |
|
|
|
Figure 17-8. Elimination of anesthetic gases is defined as the ratio of end-tidal anesthetic concentration (FA) to the last FA during administration and immediately before the beginning of elimination (FA0). During the 120-minute period after ending the anesthetic delivery, the elimination of sevoflurane and desflurane is 2 to 2.5 times faster than isoflurane or halothane (note logarithmic scale for the ordinate). (Adapted from Yasuda N, Lockhart SH, Eger EI II, et al: Comparison of kinetics of sevoflurane and isoflurane in humans. Anesth Analg 1991; 72: 316; and Yasuda N, Lockhart SH, Eger EI II, et al: Kinetics of desflurane, isoflurane, and halothane in humans. Anesthesiology 1991; 74: 489.) |
|
|
|
Figure 17-9. Both solubility and duration of anesthesia affect the decrease of the alveolar concentration (FA) from its value immediately preceding the cessation of anesthetic administration (FA0). A longer anesthetic time (from 15 minutes to 240 minutes) only slightly slows the decrease with low-soluble anesthetics (left graph). An agent with a higher blood and tissue solubility (right graph) slows the elimination of the anesthetic and enhances the effect of duration. (Adapted from Stoelting RK, Eger EI II: The effects of ventilation and anesthetic solubility on recovery from anesthesia: An in vivo and analog analysis before and after equilibrium. Anesthesiology 1969; 30: 290.) |
There are two major pharmacokinetic differences between recovery and induction. First, whereas overpressurization can increase the speed of induction, there is no “underpressurization.” Both induction and recovery rates depend on the PA to Pv gradient, and PA can never fall below zero. Second, whereas all tissues begin induction with zero anesthetic, each begins recovery with quite different anesthetic concentrations. The VRG tissues begin recovery with the same anesthetic partial pressure as that in alveoli, sincePCNS = Pblood = Palveoli. The partial pressures in muscle and fat depend on the inspired concentration during anesthesia, the duration of administration, and the anesthetic tissue solubilities. As long as an arterial-to-tissue partial pressure gradient exists, these tissues will absorb anesthetic—especially fat, since it is a huge potential reservoir whose anesthetic partial pressures are typically low after hours of anesthesia. After discontinuation of anesthesia, muscle and fat may continue to absorb anesthetic, even hours later. The redistribution continues until blood/alveolar anesthetic partial pressure falls below tissue partial pressure. This redistribution causes the early rate of decline in alveolar anesthetic concentration during recovery to exceed its early rate of increase during induction.
Because VRG tissues are highly perfused and washout of anesthetic is mostly via elimination from these tissues early in recovery, all anesthetics, regardless of duration of administration, have approximately the same rate of elimination to 50% of FA0. Unfortunately, halving the CNS concentration of anesthetic is rarely sufficient for waking the patient. More commonly, 80% to 90% of inhaled anesthetic must be eliminated before emergence. At these amounts of washout, the more soluble anesthetics are eliminated more slowly than less soluble agents.
Diffusion Hypoxia
During recovery from anesthesia, washout of high concentrations of nitrous oxide can lower alveolar concentrations of oxygen and carbon dioxide, a phenomenon called diffusion hypoxia. The resulting alveolar hypoxia can cause hypoxemia, and alveolar hypocarbia can depress respiratory drive, which may exacerbate hypoxemia. It is therefore appropriate to initiate recovery from nitrous oxide anesthesia with 100% oxygen rather than less concentrated O2/air mixtures.
P.423
Clinical Overview of Current Inhaled Anesthetics
Isoflurane
Isoflurane is a halogenated methyl ethyl ether that is a clear, nonflammable liquid at room temperature and has a high degree of pungency. It is the most potent of the volatile anesthetics in clinical use, has great physical stability, and undergoes essentially no deterioration during storage for up to 5 years or on exposure to sunlight. It has become the “gold standard” anesthetic since its introduction in the 1970s. There was a brief period of controversy concerning the use of isoflurane in patients with coronary disease because of the possibility for coronary “steal” arising from the potent effects of isoflurane on coronary vasodilation. In clinical use, however, this has been, at most, a rare occurrence.
Desflurane
Desflurane is a fluorinated methyl ethyl ether that differs from isoflurane by just one atom: a fluorine atom is substituted for a chlorine atom on the α-ethyl component of isoflurane (Fig. 17-1). The process of complete fluorination of the ether molecule has several effects. It decreases blood and tissue solubility (the blood:gas solubility of desflurane equals that of nitrous oxide), and it results in a loss of potency (the MAC of desflurane is 5 times higher than isoflurane). Moreover, fluorination of the methyl ether molecule results in a high vapor pressure owing to decreased intermolecular attraction. Thus, a new vaporizer technology was developed to deliver a regulated concentration of desflurane as a gas. It is a heated, pressurized vaporizer requiring electrical power and more frequent servicing. One of the advantages of desflurane is the near-absent metabolism to serum trifluoroacetate. This makes immune-mediated hepatitis a rare occurrence. Desflurane is the most pungent of the volatile anesthetics, and if administered via the face mask results in coughing, salivation, breath holding, and laryngospasm. In extremely dry CO2 absorbers, desflurane (and to a lesser extent isoflurane, enflurane, and sevoflurane) degrades to form carbon monoxide. Desflurane has the lowest blood:gas solubility of the potent volatile anesthetics; moreover, its fat solubility is roughly half of that of the other volatile anesthetics. Thus, desflurane requires less downward titration in long surgical procedures to achieve a rapid emergence by virtue of decreased tissue saturation. Desflurane has been associated with tachycardia, hypertension, and, in select cases, myocardial ischemia when used in high concentrations or rapidly increasing the inspired concentration (without using opioid adjuvants to prevent such a response).
Sevoflurane
Sevoflurane is a sweet-smelling, completely fluorinated methyl isopropyl ether (Fig. 17-1). Its vapor pressure is roughly one-fourth that of desflurane and it can be used in a conventional vaporizer. The blood:gas solubility of sevoflurane is second only to desflurane in terms of potent volatile anesthetics. Sevoflurane is approximately half as potent as isoflurane, and some of the preservation of potency, despite fluorination, is because of the bulky propyl side chain on the ether molecule. Sevoflurane has minimal odor, no pungency, and is a potent bronchodilator. These attributes make sevoflurane an excellent candidate for administration via the face mask on induction of anesthesia in both children and adults. Sevoflurane is half as potent a coronary vasodilator as isoflurane, but is 10 to 20 times more vulnerable to metabolism than isoflurane. The metabolism of sevoflurane results in inorganic fluoride; the increase in plasma fluoride after sevoflurane administration has not been associated with renal-concentrating defects. Unlike other potent volatile anesthetics, sevoflurane is not metabolized to trifluoroacetate; rather, it is metabolized to an acyl halide (hexafluoroisopropanol). This does not stimulate formation of antibodies.
Sevoflurane can form carbon monoxide during exposure to dry CO2 absorbents, and an exothermic reaction in dry absorbent has resulted in canister fires. New generic versions of sevoflurane have the potential to break down to hydrogen fluoride when exposed to metal compounds because of their lack of adequate water in the formulation. Sevoflurane also breaks down in the presence of the carbon dioxide absorber to form a vinyl halide called compound A. Compound A has been shown to be a dose-dependent nephrotoxin in rats, but has not been associated with renal injury in human volunteers or patients, with or without renal impairment, even when fresh gas flows are 1 L/min or less.
Xenon
Xenon is an inert gas. Difficult to obtain, and hence extremely expensive, it has received considerable interest in the last few years because it has many characteristics approaching those of an “ideal” inhaled anesthetic,4,5 although it can trigger malignant hyperthermia. Its blood:gas partition coefficient is 0.14, and unlike the other potent volatile anesthetics (except methoxyflurane), xenon provides some degree of analgesia. The MAC of xenon in humans is 71%, which might prove to be a limitation. It is nonexplosive, nonpungent, and odorless, and thus can be inhaled with ease. In addition, it does not produce significant myocardial depression.4 Because of its scarcity and high cost, new anesthetic systems need to be developed to provide for recycling of xenon. If this proves to be too difficult from either a technical or patient safety standpoint, it may be necessary to use it in a very low, or closed, fresh gas flow system to reduce wastage.
Nitrous Oxide
Nitrous oxide is a sweet-smelling, nonflammable gas of low potency (MAC = 104%) and is relatively insoluble in blood. It is most commonly administered as an anesthetic adjuvant in combination with opioids or volatile anesthetics during the conduct of general anesthesia. Although not flammable, nitrous oxide will support combustion. Unlike the potent volatile anesthetics in clinical use, nitrous oxide does not produce significant skeletal muscle relaxation, but it does have documented analgesic effects. Despite a long track record of use, controversy has surrounded nitrous oxide in four areas: its role in postoperative nausea and vomiting, its potential toxic effects on cell function via inactivation of vitamin B12, its adverse effects related to absorption and expansion into air-filled structures and bubbles, and lastly, its effect on embryonic development. The one concern that seems most valid and most clinically relevant is the ability of nitrous oxide to expand air-filled spaces because of its greater solubility in blood compared to nitrogen. Several closed gas spaces, such as the bowel and middle ear, exist in the body and other spaces may occur as a result of disease or surgery, such as a pneumothorax. Because nitrogen in air-filled spaces cannot be removed readily via the bloodstream, nitrous oxide delivered to a patient diffuses from
P.424
the blood into these closed gas spaces quite easily. Movement of nitrous oxide into these spaces continues until the partial pressure equals that of the blood and alveoli. Compliant spaces will continue to expand until sufficient pressure is generated to oppose further nitrous oxide flow into the space. The higher the inspired concentration of nitrous oxide, the higher the partial pressure required for equilibration.
Seventy-five percent nitrous oxide can expand a pneumo-thorax to double or triple its size in 10 and 30 minutes, respectively. Air-filled cuffs of pulmonary artery catheters and endotracheal tubes also expand with the use of nitrous oxide, possibly causing tissue damage via increased pressure in the pulmonary artery or trachea, respectively.6,7 In a rabbit model, the volume of an air embolus resulting in cardiovascular compromise is less during coadministration of nitrous oxide.8 Accumulation of nitrous oxide in the middle ear can diminish hearing postoperatively9 and is relatively contraindicated for tympanoplasty because the increased pressure can dislodge a tympanic graft.
Neuropharmacology of Inhaled Anesthetics
Minimum Alveolar Concentration
The pharmacodynamic effects of inhaled anesthetics must be based on a dose, and this dose is the minimum alveolar concentration or MAC. MAC is the alveolar concentration of an anesthetic at one atmosphere that prevents movement in response to a surgical stimulus in 50% of patients. It is analogous to the ED50 expressed for intravenous drugs. A variety of surgical stimuli have been used to establish the MAC for each inhaled anesthetic, but the classic, defining, noxious stimulus is incision of the abdomen. Likewise, skeletal muscle movement is the defining patient response, but other responses have been used to establish MAC as well. Experimentally determined MAC values for humans for the inhaled anesthetics are shown in Table 17-1.
The 95% confidence ranges for MAC are approximately ±25% of the listed MAC values. Manufacturer's recommendations and clinical experience establish 1.2 to 1.3 times MAC as a dose that consistently prevents patient movement during surgical stimuli. Loss of consciousness typically precedes the absence of stimulus-induced movement by a wide margin. Although 1.2 to 1.3 MAC values do not absolutely ensure the defining criteria for brain anesthesia (the absence of self-awareness and recall), vast clinical experience suggests it is extremely unlikely for a patient to be aware of, or to recall, the surgical incision at these anesthetic concentrations unless other conditions exist such that MAC is increased in that patient (Table 17-4).
Concentrations of inhaled anesthetics that provide loss of self-awareness and recall are about 0.4 to 0.5 MAC. Several lines of reasoning lead to this conclusion. First, most patients receiving only 50% nitrous oxide (approximately 0.4 to 0.5 MAC) as in a typical dentist's office will have no recall of their procedure during N2O administration. Second, various studies have shown that a shift in electroencephalogram (EEG) dominance to the anterior leads, that is, the shift from self-aware to nonself-aware, accompanies loss of consciousness, and in primates, the EEG shift and loss of consciousness occur at 0.5 MAC.10 Third, in dogs, loss of consciousness accompanies a sudden nonlinear fall in cerebral metabolic rate (CMR) at approximately 0.5 MAC (Fig. 17-10).
|
Table 17-4 Factors that Increase Minimum Alveolar Concentration |
|
|
|
|
|
|
|
Figure 17-10. The effects of halothane on cerebral metabolic rate of oxygen consumption (CMRO2) as a percentage of control (“awake”). CMRO2 is plotted versus end-tidal isoflurane concentration. Regression lines for changes in CMRO2 are drawn for each electroencephalogram-determined area. The pattern depicted here is characteristic of all of the anesthetics examined (enflurane, halothane, and isoflurane). MAC, minimum alveolar concentration. (Adapted from Stullken EH Jr, Milde JH, Michenfelder JD, et al: The nonlinear responses of cerebral metabolism to low concentrations of halothane, enflurane, isoflurane and thiopental. Anesthesiology 1977; 46: 28.) |
MAC values can be established for any measurable response. MAC-awake, or the alveolar concentration of anesthetic at which a patient opens his or her eyes to command, varies from 0.15 to 0.5 MAC.11 Interestingly, transition from awake to unconscious and back typically shows some hysteresis in that it quite consistently takes 0.4 to 0.5 MAC to lose consciousness, but less than that (as low as 0.15 MAC) to regain consciousness. This may be because of the speed of alveolar wash-in versus wash-out.12 MAC-BAR, or the alveolar concentration of anesthetic that blunts adrenergic responses to noxious stimuli, has likewise been established and is approximately 50% higher than standard MAC.13 MAC also has been established for discreet levels of EEG activity, such as onset of burst suppression or isoelectricity.
Standard MAC values are roughly additive. Administering 0.5 MAC of a potent agent and 0.5 MAC of nitrous oxide is equivalent to 1 MAC of potent agent in terms of preventing patient movement, although this does not hold over the entire range of N2O doses. MAC effects for other response parameters are not necessarily additive. Because MAC-movement probably differs from MAC for various secondary side effects (such hypothetical situations as “MAC-dysrhythmia,” “MAC-hypotension,” or “MAC-tachycardia”), combinations of a potent agent and nitrous oxide may decrease or increase these secondary effects relative to potent agent alone. For example, combining 0.6 MAC of nitrous oxide with 0.6 MAC of isoflurane produces less hypotension than 1.2 MAC of isoflurane alone because isoflurane is a more potent vasodilator and myocardial depressant at equivalent MAC than N2O.
P.425
|
Table 17-5 Factors that Decrease Minimum Alveolar Concentration |
||
|
Various factors increase (Table 17-4) or decrease (Table 17-5) MAC. Unfortunately, no single mechanism explains these alterations in MAC, supporting the view that anesthesia is the net result of numerous and widely varying physiologic alterations. In general, those factors that increase CNS metabolic activity and neurotransmission, increase CNS neurotransmitter levels, and up-regulate of CNS responses to chronically depressed neurotransmitter levels (as in chronic alcoholism) also seem to increase MAC. Conversely, those factors that decrease CNS metabolic activity, neurotransmission, and CNS neurotransmitter levels, and down-regulate CNS responses to chronically elevated neurotransmitter levels seem to decrease MAC. Many notable factors do not alter MAC, including duration of inhaled anesthetic administration, gender, type of surgical stimulation, thyroid function, hypo- or hypercarbia, metabolic alkalosis, hyperkalemia, and magnesium levels. However, there may be a genetic component influencing MAC. Red-haired females have a 19% increase in MAC compared with dark-haired females.14 These data suggest involvement of mutations of the MCIR allele. Variants of the MCIR allele also have been implicated in altering analgesic responses to a κ opioid.15 MAC also can vary in relationship to genotype and chromosomal substitutions as shown in rats.16
The Effect of Age on MAC
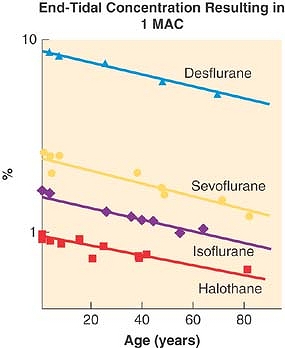
The MAC for each of the potent anesthetic gases shows a clear, age-related change (Fig. 17-11). MAC decreases with age and there are similarities between agents in the decline in MAC and age. Excluding data in patients <1 year of age (where MAC can be lower17), there is a linear model that describes a change in MAC of approximately 6% per decade, a 22% decrease in MAC from age 40 to age 80, and a 27% decrease in MAC from age 1 to 40 years.18
|
|
|
Figure 17-11. Effect of age on minimum alveolar concentration (MAC) is plotted. Regression lines are fitted to published values from separate studies. Data are from patients ages 1 to 80 years. (Adapted from Mapleson WW: Effect of age on MAC in humans: a meta-analysis. Br J Anaesth 1996; 76: 179.) |
Other Alterations in Neurophysiology
The three current, widely used, potent agents—isoflurane, desflurane, and sevoflurane—all have reasonably similar effects on a wide range of parameters including cerebral metabolic rate, the EEG, cerebral blood flow (CBF), and flow–metabolism coupling. There are notable differences in effects on ICP, cerebrospinal fluid (CSF) production and resorption, CO2 vasoreactivity, CBF autoregulation, and cerebral protection. Nitrous oxide departs from the potent agents in several important respects, and is therefore discussed separately.
Cerebral Metabolic Rate and Electroencephalogram
All of the potent agents depress CMR to varying degrees in a nonlinear fashion. Once spontaneous cortical neuronal activity is absent (an isoelectric EEG), no further decreases in CMR are generated.
Isoflurane causes a larger MAC-dependent depression of CMR than halothane. Because of this greater depression in neuronal activity, isoflurane abolishes EEG activity at doses used clinically and can usually be tolerated from a hemodynamic standpoint.19 Desflurane and sevoflurane both cause decreases in CMR similar to isoflurane.20,21 Interestingly, while both desflurane and sevoflurane depress the EEG and abolish activity at clinically tolerated doses of approximately 2 MAC,20,21 in dogs desflurane-induced isoelectric EEG reverts to continuous activity with time despite an unchanging MAC, a property unique to desflurane.21
At normal CO2 and blood pressure, no evidence of sevoflurane cerebral toxicity exists.22 With extreme hyperventilation to decrease cerebral blood flow by half, brain lactate levels increase, but significantly less than with halothane. There are conflicting data as to whether sevoflurane has a proconvulsant effect.20,23 High, long-lasting concentrations of sevoflurane (1.5 to 2.0 MAC), a sudden increase in cerebral sevoflurane
P.426
concentrations, and hypocapnia can trigger EEG abnormalities that often are associated with increases in heart rate in both adults and children.24,25 This has raised the question as to the appropriateness of sevoflurane in patients with epilepsy.26
|
|
|
Figure 17-12. Cerebral blood flow (and velocity) measured in the presence of normocapnia and in the absence of surgical stimulation in volunteers receiving halothane or isoflurane. At light levels of anesthesia, halothane (but not isoflurane) increased cerebral blood flow. At 1.6 minimum alveolar concentration (MAC), isoflurane also increased cerebral blood flow. (Adapted from Eger EI II: Isoflurane (Forane): A compendium and reference. Madison, Ohio Medical Products, 1985.) Cerebral blood flow velocity measured before and during sevoflurane and desflurane anesthesia up to 1.5 MAC showed no change in cerebral blood flow and velocity. (Adapted from Bedforth NM, Hardman JG, Nathanson MH: Cerebral hemodynamic response to the introduction of desflurane: A comparison with sevoflurane. Anesth Analg 2000; 91: 152.) |
Cerebral Blood Flow, Flow–Metabolism Coupling, and Autoregulation
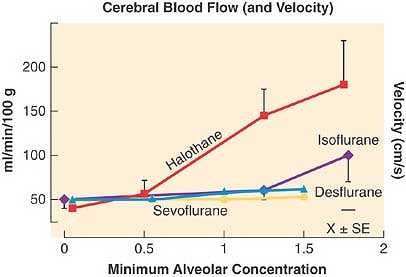
All of the potent agents increase CBF in a dose-dependent manner. Isoflurane, sevoflurane, and desflurane cause far less cerebral vasodilation per MAC-multiple than halothane (Fig. 17-12). In human studies, isoflurane produces insignificant or no changes in CBF.27 Desflurane and sevoflurane both influence CBF in a fashion similar to isoflurane.20,21 All of these inhaled anesthetic agents affect CBF in a time-dependent as well as dose-dependent manner. In animals, an initial dose-dependent increase in CBF with halothane and isoflurane administration recovers to preinduction levels approximately 2 to 5 hours after induction. The mechanism of this recovery is unclear.
The increase in CBF with increasing dose caused by the potent agents occurs despite decreases in CMR. This phenomenon has been called uncoupling, but from a mechanistic standpoint, true uncoupling of flow from metabolism may not occur. That is, as CMR is depressed by the volatile anesthetics, there still is a coupled decline in CBF opposed by a coincident direct vasodilatory effect on the cerebral blood vessels. The net effect on the cerebral vessels depends on the sum of indirect vasoconstricting and direct vasodilating influences.
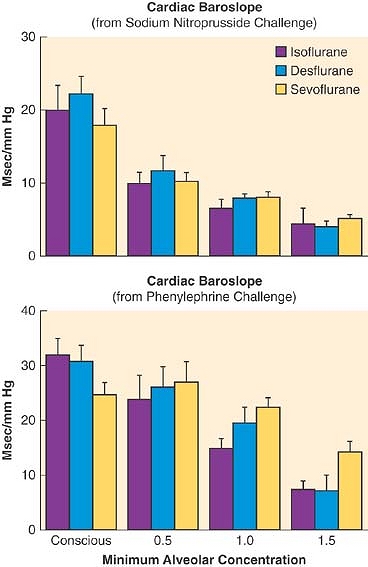
Autoregulation is the intrinsic myogenic regulation of vascular tone. In normal brain, the mechanisms of autoregulation of CBF over a range of mean arterial pressures from 50 to 150 mm Hg are incompletely understood. Because the volatile anesthetics are direct vasodilators, all are considered to diminish autoregulation in a dose-dependent fashion such that at high anesthetic doses CBF is essentially pressure-passive. Sevoflurane preserves autoregulation up to approximately 1 MAC.20 At 1.5 MAC, the dynamic rate of autoregulation (change in middle cerebral artery blood flow after a rapid transient decrease in blood pressure) is better preserved with sevoflurane than isoflurane (Fig. 17-13). This may be a result of less of a direct vasodilator effect of sevoflurane, preserving the ability of the vessel to respond to changes in blood pressure at 1.5 MAC. Based on a similar model but a separate study of dynamic autoregulation of cerebral blood flow, 0.5 MAC desflurane reduced autoregulation and isoflurane did not. At 1.5 MAC, both anesthetics substantially reduced autoregulation (Fig. 17-13).
|
|
|
Figure 17-13. Dynamic rate of autoregulation (the change in middle cerebral artery blood flow after a rapid transient decrease in blood pressure) during awake (or fentanyl and N2O baseline), 0.5, and 1.5 minimum alveolar anesthetic concentration (MAC) anesthesia. Values are mean ± SE (SD for sevoflurane). *P < 0.05 versus baseline, **P < 0.001 versus baseline and sevoflurane. (Adapted from Summors AC, Gupta AK, Matta BF: Dynamic cerebral autoregulation during sevoflurane anesthesia: A comparison with isoflurane. Anesth Analg 1999; 88: 341–345; and Strebel S, Lam A, Matta B, et al: Dynamic and static cerebral autoregulation during isoflurane, desflurane, and propofol anesthesia. Anesthesiology 1995; 83: 66–76.) |
Intracerebral Pressure
Probably the area of greatest clinical interest to the anesthesiologist is the effect of volatile anesthesia on intracerebral pressure (ICP). In general, ICP will increase or decrease in proportion to changes in CBF. Isoflurane increases ICP minimally in animals both with and without brain pathology, including those with an already elevated ICP.28 In human studies there usually are mild increases in ICP with isoflurane administration that are blocked or blunted by hyperventilation or barbiturate coadministration.29 There are some contradictory data, however. In one human study, hypocapnia did not prevent elevations in ICP with isoflurane administration in patients with space-occupying brain lesions.30 However, isoflurane-induced increases in ICP tend to be of short duration, in one study only 30 minutes.31
Like isoflurane, both sevoflurane and desflurane >1 MAC produce mild increases in ICP, paralleling their mild increases in CBF.20,21,32,33 One potential advantage of sevoflurane is that its lower pungency and airway irritation may lessen the risk of coughing and bucking and the associated rise in ICP as compared with desflurane or isoflurane. In fact, introduction of desflurane after propofol induction of anesthesia has led to significant increases in heart rate, mean arterial pressure, and middle cerebral artery blood flow velocity that were not noted in patients given sevoflurane.34 This may relate to the airway irritant effects of desflurane rather than a specific alteration in
P.427
neurophysiology. However, several studies in both children and adults suggest that increases in ICP from desflurane are slightly greater than from either isoflurane or sevoflurane.35,36The bottom line is that all three potent agents may be used at appropriate doses, especially with adjunctive and compensatory therapies, in just about any neurosurgical procedure.
Cerebrospinal Fluid Production and Resorption
Isoflurane does not appear to alter CSF production,31 but may increase, decrease, or leave unchanged the resistance to resorption depending on dose. Sevoflurane at 1 MAC depresses CSF production up to 40%.37 Desflurane at 1 MAC leaves CSF production unchanged or increased.35,38 In general, anesthetic effects on ICP via changes in CSF dynamics are clinically far less important than anesthetic effects on CBF.
Cerebral Blood Flow Response to Hypercarbia and Hypocarbia
Significant hypercapnia is associated with dramatic increases in CBF whether or not volatile anesthetics are administered. As discussed earlier, hypocapnia can blunt or abolish volatile anesthetic-induced increases in CBF depending on when the hypocapnia is produced. This vasoreactivity to CO2 may be somewhat altered by the volatile anesthetics as compared with normal. CO2 vasoreactivity under desflurane anesthesia is normal up to 1.5 MAC,28 and CO2 vasoreactivity for sevoflurane is preserved at 1 MAC.39
Cerebral Protection
In one study, cerebral hypoperfusion secondary to hypotension from isoflurane was associated with better tissue oxygen content than during hypotension by other means, consistent with the profound decrease in cerebral metabolic rate of oxygen consumption (CMRO2) seen with isoflurane.40 Both sevoflurane and desflurane have been shown to improve neurologic outcome in comparison to N2O-fentanyl after incomplete cerebral ischemia in a rat model.41,42 In piglets undergoing low-flow cardiopulmonary bypass, desflurane improved neurologic outcome compared with a fentanyl/droperidol-based anesthetic.43 In humans, desflurane has been shown to increase brain tissue PO2 during administration, and to maintain PO2 to a greater extent than thiopental during temporary cerebral artery occlusion during cerebrovascular surgery.44 Human neuroprotection outcome studies for sevoflurane and desflurane have not been published.
Processed Electroencephalograms and Neuromonitoring
All of the volatile anesthetics produce dose-dependent effects on the EEG, sensory-evoked potentials (SEPs) and motor-evoked potentials (MEPs). EEGs recorded on the scalp can be processed to quantify the amount of activity in each of four frequency bands: delta (0 to 3 Hz), theta (4 to 7 Hz), alpha (8 to 13 Hz), and beta (>13 Hz). All three currently used agents at <1 MAC and N2O at 30 to 70% can produce shifts to increasing frequencies. Between 1 and 2 MAC the potent agents produce shifts to decreasing frequencies and increases in amplitude. At >2 MAC, all of the potent agents can produce burst suppression or electrical silence. These are important factors to remember because EEG changes during administration of general anesthesia can also be caused by hypoxia, hypercarbia, and hypothermia. The EEG must always be interpreted within the appropriate clinical context.
All of the volatile agents cause a dose-dependent increase in latency and decrease in amplitude in all cortical SEP modalities. In subcortical modalities, such as brainstem auditory evoked potentials, these agents are associated with negligible effects. In general, visual evoked potentials are somewhat more sensitive to the effects of the volatile anesthetics than somatosensory evoked potentials. Like EEGs, these effects from anesthetics must be kept in mind when changes during SEPs occur, and appropriate doses of the volatile agents must be used. Sudden changes in the anesthetic regimen (>0.5 MAC) also seem to have greater effects on SEPs than more gradual changes.
MEPs evaluate the functional integrity of descending motor pathways. The evoked response is most commonly recorded as a muscle potential or a peripheral nerve signal. The trigger is typically transosseous activation via electrical or magnetic stimulation. MEPs are exquisitely sensitive to depression by volatile anesthetics, which are usually avoided in these cases.
Nitrous Oxide
The effects of nitrous oxide on cerebral physiology are not clear. Both the MAC for N2O and its effects on CMR vary widely depending on species. The difference in CMR effects may in part be accounted for by differences in MAC, but MAC-equivalent effects on CMR also differ. Several studies in dogs, goats, and swine found that N2O increases CMRO2 and CBF, while in rodents no such increases or only slight increases occur. In human studies, N2O administration preserved CBF but decreased CMRO2.19
Another problem is the fact that N2O is a coanesthetic used to supplement potent agents, not a complete anesthetic in itself, and CMR effects may differ depending on presence or absence of potent agent as well as the particular agent and dose. Addition of N2O to 1 or 2.2 MAC isoflurane does not alter CMRO2, but it does increase CBF at 1 MAC but not 2.2 MAC.
Barbiturates, narcotics, or a combination of the two appear to decrease or eliminate the increases in CMR and CBF produced by N2O. The effect of pentobarbital/N2O is dose-dependent, with preserved increases in CMR by N2O at low-dose pentobarbital, and no changes in CMR at high-dose pentobarbital.45 N2O and benzodiazepine coadministration is particularly confusing. Midazolam/N2O in dogs increased CBF but did not alter CMRO2,46 while the opposite was true in rats,47 and both CBF and CMRO2 declined in rats given diazepam/N2O. N2O administration increases ICP, but as is the case for CMR and CBF, changes in ICP are decreased or eliminated by a variety of coanesthetics and, more importantly, by hypocapnia.
N2O appears to have an antineuroprotective effect, as addition of N2O to isoflurane during temporary ischemia is associated with greater tissue damage and worsened neurologic outcome.47 In a study in mice, survival time after a hypoxic event was decreased by addition of N2O.48 Given the conflicting data on the effects of N2O on CMR, CBF, ICP, and the apparent antineuroprotective effect of this agent, avoidance or discontinuation of its use should be considered in surgical cases with a high likelihood of elevated ICP or significant cerebral ischemia.
The Circulatory System
Hemodynamics
The cardiac, vascular, and autonomic effects of the volatile anesthetics have been carefully defined through a number of studies carried out in human volunteers not undergoing surgery.49,50,51,52,53,54 In general, the information from these volunteer
P.428
studies has translated well to the patient population commonly exposed to these anesthetics during elective and emergent surgeries.
|
|
|
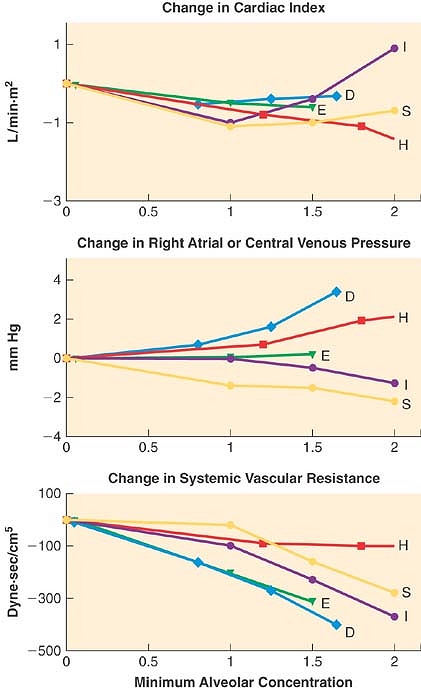
Figure 17-14. Heart rate and blood pressure changes (from awake baseline) in volunteers receiving general anesthesia with halothane (H), enflurane (E), isoflurane (I), desflurane (D), or sevoflurane (S). Halothane and sevoflurane produced little or no change in heart rate at <1.5 minimum alveolar concentration. All anesthetics caused similar decreases in blood pressure. (Adapted from Malan TP Jr, DiNardo JA, Isner RJ, et al: Cardiovascular effects of sevoflurane compared with those of isoflurane in volunteers. Anesthesiology 1995; 83: 918; Weiskopf RB, Cahalan MK, Eger EI II, et al: Cardiovascular actions of desflurane in normocarbic volunteers. Anesth Analg 1991; 73: 143; and Calverley RK, Smith NT, Prys-Roberts C, et al: Cardiovascular effects of enflurane anesthesia during controlled ventilation in man. Anesth Analg 1978; 57: 619.) |
A common effect of the potent volatile anesthetics has been a dose-related decrease in arterial blood pressure, with essentially no differences noted between the volatile anesthetics at steady-state, equianesthetic concentrations (Fig. 17-14). Their primary mechanism to decrease blood pressure with increasing dose is related to their potent effects to lower regional and systemic vascular resistance (Fig. 17-15).
In volunteers, sevoflurane up to about 1 MAC results in minimal, if any, changes in steady-state heart rate while enflurane, isoflurane, and desflurane increase it 5 to 10% from baseline (Fig. 17-14). Both desflurane and, to a lesser extent, isoflurane have been associated with transient and significant increases in heart rate during rapid increases in the inspired concentration of either anesthetic.55,56 The mechanism(s) underlying these transient heart rate surges is likely due to the relative pungency of these anesthetics, which stimulates airway receptors to elicit a reflex tachycardia.57 The tachycardia can be lessened with fentanyl, alfentanil, or clonidine pretreatment.58,59,60
|
|
|
Figure 17-15. Cardiac index, central venous pressure (or right atrial pressure), and systemic vascular resistance changes (from awake baseline) in volunteers receiving general anesthesia with halothane (H), enflurane (E), isoflurane (I), desflurane (D), or sevoflurane (S). Increases in central venous pressure from halothane and desflurane might be due to different mechanisms. With halothane, the increase might be due to myocardial depression, whereas with desflurane, the increase is more likely due to venoconstriction. (Adapted from Malan TP Jr, DiNardo JA, Isner RJ, et al: Cardiovascular effects of sevoflurane compared with those of isoflurane in volunteers. Anesthesiology 1995; 83: 918; Weiskopf RB, Cahalan MK, Eger EI II, et al: Cardiovascular actions of desflurane in normocarbic volunteers. Anesth Analg 1991; 73: 143; and Calverley RK, Smith NT, Prys-Roberts C, et al: Cardiovascular effects of enflurane anesthesia during controlled ventilation in man. Anesth Analg 1978; 57: 619.) |
Myocardial Contractility
Myocardial contractility indices have been directly evaluated in animals and indirectly evaluated in humans during the administration of each of the volatile anesthetics. Human studies with isoflurane, sevoflurane, and desflurane have not demonstrated significant changes in echocardiographic-determined indices of myocardial function, including the more noteworthy measurement of the velocity of circumferential fiber shortening (Fig. 17-16). More precise indices of myocardial contractility have been obtained for sevoflurane, isoflurane, and desflurane in chronically instrumented dogs
P.429
after autonomic denervation of the heart. Isoflurane, desflurane, and sevoflurane resulted in a dose-dependent depression of myocardial function with no differences between the three anesthetics (Fig. 17-17).
|
|
|
Figure 17-16. Noninvasive assessment of myocardial contractility with echocardiography during anesthesia in volunteers. Sevoflurane, desflurane, and isoflurane did not cause changes suggestive of myocardial depression. (Adapted from Malan TP Jr, DiNardo JA, Isner RJ, et al: Cardiovascular effects of sevoflurane compared with those of isoflurane in volunteers. Anesthesiology 1995; 83: 918; Weiskopf RB, Cahalan MK, Eger EI II, et al: Cardiovascular actions of desflurane in normocarbic volunteers. Anesth Analg 1991; 73: 143; and Calverley RK, Smith NT, Prys-Roberts C, et al: Cardiovascular effects of enflurane anesthesia during controlled ventilation in man. Anesth Analg 1978; 57: 619.) |
Other Circulatory Effects
Most of the volatile anesthetics have been studied during both controlled and spontaneous ventilation.51,61,62 The process of spontaneous ventilation reduces the high intrathoracic pressures from positive pressure ventilation. The negative intrathoracic pressure during the inspiratory phase of spontaneous ventilation augments venous return and cardiac filling and improves cardiac output and, hence, blood pressure. Spontaneous ventilation is associated with higher PaCO2, causing cerebral and systemic vascular relaxation. This contributes to an improved cardiac output via afterload reduction. Thus, spontaneous ventilation decreases systemic vascular resistance and increases heart rate, cardiac output, and stroke volume as contrasted to positive pressure ventilation. It has been suggested that spontaneous ventilation might improve the safety of inhaled anesthetic administration because the concentration of a volatile anesthetic that produces cardiovascular collapse exceeds the concentration that results in apnea.63
A curious observation with the potent volatile anesthetics has been an alteration in the cardiovascular effects during prolonged anesthetic exposures, noted as a small increase in heart rate and cardiac index, a gradual decrease in systemic vascular resistance, and no change in myocardial indices.51,52 The mechanism(s) of this effect are not clear.
Nitrous oxide is commonly combined with potent volatile anesthetics to maintain general anesthesia. Nitrous oxide has unique cardiovascular actions. It increases sympathetic nervous system activity and vascular resistance when given in a 40% concentration.49,64 When nitrous oxide is combined with volatile anesthetics and compared with equipotent concentrations of the volatile anesthetic without nitrous oxide, there is an increased systemic vascular resistance and an improved arterial pressure with little effect on cardiac output.51,65 These effects might not be due solely to sympathetic activation from nitrous oxide per se, but may be partially attributed to a decrease in the concentration of the coadministered potent volatile anesthetic required to achieve a MAC equivalent when using nitrous oxide.
Oxygen consumption is decreased approximately 10 to 15% during general anesthesia.66 The distribution of cardiac output also is altered by anesthesia. Blood flow to liver, kidneys, and gut is decreased, particularly at deep levels of anesthesia. In contrast, blood flow to the brain, muscle, and skin is increased or not changed during general anesthesia.67 In humans, increases in muscle blood flow are noted with isoflurane, desflurane, and sevoflurane with very small differences between anesthetics at equipotent concentrations.68
Isoflurane, sevoflurane, and desflurane do not predispose patients to ventricular arrhythmias, nor sensitize the heart to the arrhythmogenic effects of epinephrine (Fig. 17-18). Some of the differences between volatile anesthetics in their ability to promote other arrhythmias can be attributed to their direct effects on cardiac pacemaker cells and conduction pathways.69 Sinoatrial node discharge rate is slowed by the volatile anesthetics70 and conduction in the His-Purkinje system and
P.430
conduction pathways in the ventricle also is prolonged by the volatile anesthetics.69
|
|
|
Figure 17-17. Myocardial contractility indices from chronically instrumented dogs. For these measurements, pharmacologic blockade of the autonomic nervous system was established to eliminate neural or circulating humoral influences on the inotropic state of the heart. The conscious control data were assigned 100%, and subsequent reductions in the inotropic state are depicted for both 1 and 1.5 minimum alveolar anesthetic concentrations of sevoflurane, desflurane, and isoflurane. There were no differences between these three volatile anesthetics. Mw, slope of the regional preload recruitable stroke work relationship; dP/dt50, change in pressure per unit of time. (Adapted from Pagel PS, Kampine JP, Schmeling WT, et al: Influence of volatile anesthetics on myocardial contractility in vivo: Desflurane versus isoflurane. Anesthesiology 1991; 74: 900; and Harkin CP, Pagel PS, Kersten JR, et al: Direct negative inotropic and lusitropic effects of sevoflurane. Anesthesiology 1994; 81: 156.) |
|
|
|
Figure 17-18. The dose of epinephrine associated with cardiac arrhythmias in animal and human models was least with halothane. The ether anesthetics—isoflurane, desflurane, and sevoflurane—required three- to sixfold greater doses of epinephrine to cause arrhythmias. (Adapted from Navarro R, Weiskopf RB, Moore MA, et al: Humans anesthetized with sevoflurane or isoflurane have similar arrhythmic response to epinephrine. Anesthesiology 1994; 80: 545; Weiskopf RB, Eger EI II, Holmes MA, et al: Epinephrine-induced premature ventricular contractions and changes in arterial blood pressure and heart rate during I-653, isoflurane, and halothane anesthesia in swine. Anesthesiology 1989; 70: 293; Hayashi Y, Sumikawa K, Tashiro C, et al: Arrhythmogenic threshold of epinephrine during sevoflurane, enflurane, and isoflurane anesthesia in dogs. Anesthesiology 1988; 69: 145; and Moore MA, Weiskopf RB, Eger EI II, et al: Arrhythmogenic doses of epinephrine are similar during desflurane or isoflurane anesthesia in humans. Anesthesiology 1993; 79: 943.) |
Coronary Steal
Isoflurane (and most other potent volatile anesthetics) increases coronary blood flow many times beyond that of the myocardial oxygen demand, thereby creating potential for “steal.” Steal is the diversion of blood from a myocardial bed with limited or inadequate perfusion to a bed with more adequate perfusion; especially one that has a remaining element of autoregulation. Work in a chronically instrumented, canine model of multivessel coronary artery obstruction, has shown that neither isoflurane, sevoflurane, or desflurane at concentrations up to 1.5 MAC resulted in abnormal collateral coronary blood flow redistribution (steal), whereas adenosine, a potent coronary vasodilator, clearly resulted in abnormal flow distribution.71,72,73,74 Interestingly, sevoflurane favorably increased (rather than decreased) collateral coronary blood flow in this instrumented animal model when aortic pressure was held constant by pharmacologic support of blood pressure.73
Myocardial Ischemia and Cardiac Outcome
Not surprisingly, the clinical relevance of coronary steal with isoflurane has been debated and is generally thought to be minimal.75 Outcome studies have failed to associate the use of isoflurane in patients undergoing coronary artery bypass operations with an increased incidence of myocardial infarction or perioperative death.75,76,77 Most studies would suggest that determinants of myocardial oxygen supply and demand, rather than the anesthetic, are of far greater importance to patient outcomes.
Several studies have evaluated sevoflurane and desflurane in reference to comparator anesthetics, in terms of myocardial ischemia and outcome in patients with coronary artery disease either undergoing noncardiac or coronary artery bypass graft surgery.78,79 In both populations, sevoflurane appears to be essentially equivalent to isoflurane in terms of the incidence of myocardial ischemia and adverse cardiac outcomes. Desflurane appears to result in similar outcome effects as isoflurane in cardiac patients having coronary artery bypass grafting80 with one exception. In a study in which desflurane was given without opioids to patients with coronary artery disease requiring coronary artery bypass graft surgery, significant ischemia mandating the use of beta-blockers was noted.81 Desflurane has not been evaluated in terms of ischemia and outcome in a patient population with coronary disease undergoing noncardiac surgery.
Cardioprotection from Volatile Anesthetics
A preconditioning stimulus such as brief coronary occlusion and ischemia initiates a signaling cascade of intracellular events that reduces ischemia and reperfusion myocardial injury. There is a memory effect from an ischemic stimulus that offers 2 to 3 hours of protection. The volatile anesthetics mimic ischemic preconditioning and trigger a similar cascade of intracellular events resulting in myocardial protection that lasts beyond the elimination of the anesthetic.82 Numerous factors may be involved in preconditioning, including the sodium:hydrogen exchanger, the adenosine receptor (particularly α1 and α2 subtypes), inhibitory G proteins, protein kinase C, tyrosine kinase, and potassium (KATP) channel opening. Pharmacologic blockade of these factors (e.g., with adenosine blockers, delta 1 opioids, pertussis toxin, or glibenclamide) reduces or eliminates the cardioprotective effect of ischemic or volatile anesthetic preconditioning.82,83 Alternatively, administration of certain drugs can mimic ischemic or volatile anesthetic preconditioning. These include adenosine, opioid agonists, and KATP channel openers. In contrast to the inhalation of volatile anesthetics, these cardioprotective drugs must be delivered into a coronary artery because systemic administration can have serious side effects.
Lipophilic volatile anesthetics diffuse through myocardial cell membranes and alter mitochondrial electron transport, leading to reactive oxygen species formation.83 This may be the trigger for preconditioning via protein kinase C activation of KATP channel opening.84,85 Approximately 30 to 40% of the cardioprotection from the volatile anesthetics appears to be related to a reduced loading of calcium into the myocardial cells during ischemia. Preconditioned hearts may tolerate ischemia for 10 minutes longer than nonconditioned hearts.86
While these evolving data generally derive from animal models, there now is increasing evidence in cardiac patient populations that anesthetic cardioprotection lessens myocardial damage (based on troponin levels) during “on and off pump” cardiac surgery.87,88 This effect seems to be common to all current potent volatile anesthetics, and may favorably influence intensive care unit length of stay after coronary surgery.89 Sulfonylurea oral hyperglycemic drugs close KATP channels, abolishing anesthetic preconditioning. They should be discontinued 24 to 48 hours prior to elective surgery in high-risk patients.82 But hyperglycemia also prevents preconditioning, so insulin therapy should be started when holding oral agents.90 Recent evidence suggests that volatile anesthetics may protect other organs from ischemic injury including kidney, liver, and brain.91,92,93
P.431
|
|
|
Figure 17-19. Summary data of the baroreflex regulation of heart rate (R-R interval) in response to a decreasing pressure stimulus (sodium nitroprusside) or in response to an increasing pressure stimulus (phenylephrine). These data were acquired in healthy volunteers who were randomized to receive isoflurane, desflurane, or sevoflurane. With increasing minimum alveolar anesthetic concentration, each of the volatile anesthetics led to a progressive reduction in the cardiac baroslope (an index of baroreflex sensitivity derived by relating changes in mean pressure to changes in R-R interval). There were no statistical differences between anesthetics. (Adapted from Ebert TJ, Harkin CP, Muzi M: Cardiovascular responses to sevoflurane: A review. Anesth Analg 1995; 81: S11.) |
Autonomic Nervous System
Studies that have focused on the efferent activity of the parasympathetic and sympathetic nervous systems indicate that the volatile anesthetics depress their activity in a dose-dependent fashion.94,95 However, because the autonomic nervous system is importantly modulated by baroreceptor reflex mechanisms, the effects of the anesthetic on the efferent system cannot be reported without taking into account their effects on different components of the baroreflex arc. Thus, although both limbs of the autonomic nervous system have been shown to be attenuated by the anesthetics, the afferent activity from the arterial baroreceptors has been found to be increased with some of the anesthetics, such as isoflurane.96 This increased discharge of the baroreceptors actually contributes to the depression of the entire baroreflex arc by tonically lowering the overall level of outflow of the sympathetic nervous system. From the perspective of clinical relevance, studies have examined the behavior of the arterial baroreflex system during a hypotensive or hypertensive stimulus by evaluating changes in heart rate and sympathetic nerve activity. The arterial baroreflex is the most rapidly responding system to blood pressure perturbations. Early investigations focused primarily on the regulation of heart rate, which reflects a primarily vagal-mediated end point. Isoflurane reduces, in a dose-dependent fashion, arterial baroreflex control of heart rate.97 Similar effects on the reflex control of heart rate have recently been demonstrated with sevoflurane and desflurane (Fig. 17-19).98,99,100
There is greater difficulty in evaluating the sympathetic component of the baroreflex arc in humans, but a technique called sympathetic microneurography has been used to directly record vasoconstrictor impulses directed to blood vessels in humans.50,55 There is a dose-dependent depression of the reflex control of sympathetic outflow that appears to be relatively equivalent for isoflurane, sevoflurane, and desflurane (Fig. 17-20). Importantly, at low levels of anesthesia (e.g., 0.5 MAC), there is little if any depression of reflex function and this might have important implications in the compromised patient population. Opioid and benzodiazepine adjuvants have only minimal effects on reflex function and combining these with low levels of potent anesthetics might preserve reflex function.101,102 Another important observation has been the more rapid return of baroreflex function with the less soluble anesthetic sevoflurane versus isoflurane.103 This might add to hemodynamic stability in the postoperative period when tissue concentrations of the volatile anesthetics are declining.
Desflurane has a unique and prominent effect on sympathetic outflow in humans, which is not apparent in animal models. With increasing steady-state concentrations of desflurane, there is a progressive increase in resting sympathetic nervous system activity and plasma norepinephrine levels.50,55,104 Despite this increase in tonic sympathetic outflow, blood pressure decreases similarly to sevoflurane and isoflurane. This raises the question as to whether desflurane has the ability to uncouple neuroeffector responses. In addition, desflurane can cause marked activation of the sympathetic nervous system when the inspired concentration is increased, especially to
P.432
concentrations above 5 to 6% (Fig. 17-21).50,55,104 There is a transient surge in sympathetic outflow leading to both hypertension and tachycardia. In addition, the endocrine axis is activated as evidenced by 15- to 20-fold increases in plasma anti-diuretic hormone and epinephrine and norepinephrine (Fig. 17-22). The hemodynamic response persists for 4 to 5 minutes and the endocrine response persists for 15 to 25 minutes. Adequate concentrations of opioids or clonidine given prior to increasing the concentration of desflurane have been shown to attenuate these responses.58,59,60 The source of the neuroendocrine activation has been actively sought, and it would appear that there are receptors in both the upper and the lower airways, and/or perhaps in a highly perfused tissue near the airways, that initiates the sympathetic activation.57
|
|
|
Figure 17-20. The sympathetic baroreflex function of healthy volunteers randomized to receive isoflurane, desflurane, or sevoflurane. The slope (sensitivity) is the relationship between decreasing diastolic pressure and increasing efferent sympathetic nerve activity. The reflex regulation of sympathetic outflow was fairly well preserved at 0.5 and 1.0 minimum alveolar anesthetic concentration (MAC) of anesthetic. At 1.5 MAC, there was a 50% decrease in the slope with all anesthetics. (Adapted from Ebert TJ, Harkin CP, Muzi M: Cardiovascular responses to sevoflurane: A review. Anesth Analg 1995; 81: S11.) |
|
|
|
Figure 17-21. Consecutive measurements of sympathetic nerve activity (SNA; mean ± SE) from human volunteers during induction of anesthesia with propofol and the subsequent mask administration of sevoflurane or desflurane for a 10-minute period. The inspired concentration of these anesthetics was increased at 1-minute intervals beginning after propofol administration (0.41 MAC of sevoflurane and desflurane). In both groups, propofol reduced SNA and mean arterial pressure. Desflurane resulted in significant increases in SNA that persisted throughout the 10-minute mask administration period. (Adapted from Ebert TJ, Muzi M, Lopatka CW: Neurocirculatory responses to sevoflurane in humans. A comparison to desflurane. Anesthesiology 1995; 83: 88.) |
The Pulmonary System
General Ventilatory Effects
All volatile anesthetics decrease tidal volume and increase respiratory rate such that there are only minor effects on decreasing minute ventilation (Fig. 17-23). The ventilatory effects are dose-dependent, with higher concentrations of volatile anesthetics resulting in greater decreases in tidal volume and greater increases in respiratory rate, with the exception of isoflurane, which does not increase respiratory rate above 1 MAC. Their net effect of a gradual decrease in minute ventilation has been associated with increasing resting Paco2. The respiratory depression can be partially antagonized during surgical stimulation where respiratory rate and tidal volume have been shown to increase, resulting in a decrease in the Paco2 (Fig. 17-24). N2O increases respiratory rate as much or more than the inhaled anesthetics. When N2O is added to sevoflurane or desflurane, resting Paco2decreases relative to equi-MAC concentrations of sevoflurane or desflurane in O2. The degree of respiratory depression from inhaled anesthetics is reduced when anesthesia administration exceeds 5 hours.105
|
|
|
Figure 17-22. Stress hormone responses to a rapid increase in anesthetic concentration, from 4 to 12% inspired. Volunteers given desflurane showed a larger increase in plasma epinephrine and norepinephrine concentrations than when given isoflurane. Data are mean ± SE. A = awake value; B = value after 32 minutes of 0.55 minimum alveolar concentration; time represents minutes after the first breath of increased anesthetic concentration. (Adapted from Weiskopf RB, Moore MA, Eger EI II, et al: Rapid increase in desflurane concentration is associated with greater transient cardiovascular stimulation than with rapid increase in isoflurane concentration in humans. Anesthesiology 1994; 80: 1035.) |
Ventilatory Mechanics
FRC is decreased during general anesthesia; this has been explained by a number of mechanisms including a decrease in the intercostal muscle tone, alteration in diaphragm position,
P.433
changes in thoracic blood volume, and the onset of phasic expiratory activity of respiratory muscles. About 40% of the muscular work of breathing is via intercostal muscles and about 60% is from the diaphragm. The diaphragmatic muscle function is relatively spared when contrasted to the parasternal intercostal muscles. However, inspiratory rib cage expansion is reasonably well maintained during anesthesia because of preserved activity of the scalene muscles. Expiration is generally considered a passive function mediated by the elastic recoil of the lung. The process of applying a resistance or load to expiration typically results in a slowing of respiration, but under anesthesia, additional responses include a substantial asynchrony of the thoracic movements with respiration. This suggests that in patients with pulmonary disease associated with increased expiratory resistance, the act of spontaneous ventilation during general anesthesia might be poorly tolerated.
|
|
|
Figure 17-23. Comparison of mean changes in resting Paco2, tidal volume, respiratory rate, and minute ventilation in patients anesthetized with either halothane, isoflurane, enflurane, sevoflurane, desflurane, or nitrous oxide (N, N2O). Anesthetic-induced tachypnea compensates in part for the ventilatory depression caused by all volatile anesthetics (decrease in minute ventilation and tidal volume, and concomitant increase in Paco2). Desflurane results in the greatest increase in Paco2 with corresponding reductions in tidal volume and minute ventilation. Isoflurane, like all other inhaled agents, increases respiratory rate, but does not result in dose-dependent tachypnea. (Adapted from Lockhart SH, Rampil IJ, Yasuda N, et al: Depression of ventilation by desflurane in humans. Anesthesiology 1991; 74: 484; Doi M, Ikeda K: Respiratory effects of sevoflurane. Anesth Analg 1987; 66: 241; Fourcade HE, Stevens WC, Larson CP Jr, et al: The ventilatory effects of Forane, a new inhaled anesthetic. Anesthesiology 1971; 35: 26; and Calverley RK, Smith NT, Jones CW, et al: Ventilatory and cardiovascular effects of enflurane anesthesia during spontaneous ventilation in man. Anesth Analg 1978; 57: 610.) |
|
|
|
Figure 17-24. The effect of surgical stimulation on the ventilatory depression of inhaled anesthesia with isoflurane in the presence and absence of nitrous oxide (N2O). Surgical stimulation increased alveolar ventilation and decreased Paco2 at all depths of anesthesia examined. (Adapted from Eger EI 2nd, Dolan WM, Stevens WC, et al: Surgical stimulation antagonizes the respiratory depression produced by Forane. Anesthesiology 1972; 36: 544.) |
Response to Carbon Dioxide and Hypoxemia
In awake humans, the central chemoreceptors respond vigorously to changes in arterial carbon dioxide tension such that minute ventilation increases 3 L/min per a 1-mm Hg increase in Paco2. All of the inhaled anesthetics produce a dose-dependent depression of the ventilatory response to hypercarbia (Fig. 17-25). The addition of nitrous oxide to a volatile anesthetic had been thought to diminish Paco2 responses less than an equi-MAC dose of the anesthetic alone, however, this does not appear to be the case for desflurane (Fig. 17-25). In patients with chronic obstructive pulmonary disease, there is an impaired response to increased Paco2 under anesthesia.
The threshold at which breathing stops, called the apneic threshold, can be determined during anesthesia with spontaneous ventilation. It is generally 4 to 5 mm Hg below the prevailing resting Paco2 and unrelated to the slope of the co2 response curves or to the level of the resting Paco2. The clinical
P.434
relevance of this threshold may be recognized when assisting ventilation in an anesthetized patient who is breathing spontaneously. This only serves to lower the Paco2 to approach that of the apneic threshold, therefore mandating more control of ventilation.
|
|
|
Figure 17-25. All inhaled anesthetics produce similar dose-dependent decreases in the ventilatory response to carbon dioxide (CO2). N2O, nitrous oxide. (Adapted from Eger EI II: Desflurane. Anesth Rev 1993; 20: 87; and Doi M, Ikeda K: Respiratory effects of sevoflurane. Anesth Analg 1987; 66: 241.) |
Inhaled anesthetics, including nitrous oxide, produce a dose-dependent attenuation of the ventilatory response to hypoxia. This action appears to depend on the peripheral chemoreceptors. In fact, even subanesthetic concentrations of volatile anesthetics (0.1 MAC) elicit anywhere from a 15 to 75% depression of the ventilatory drive to hypoxia (Fig. 17-26). The mechanism of this depression still remains poorly understood. Studies have suggested that hypoxia may decrease the probability that potassium channels are open, thus causing membrane depolarization, an influx of calcium ions, and a release of neurotransmitters.106 One theory is that the potassium channels are responding to reactive oxygen species. One study found the administration of antioxidants prior to the administration of a volatile anesthetic prevented the depression of the hypoxic response.107 The extreme sensitivity of the volatile anesthetics to inhibit ventilatory responses to hypoxia has important clinical implications. Residual effects of volatile anesthetics may impair the ventilatory drive of patients in the recovery room. In this regard, the short-acting anesthetics (sevoflurane and desflurane) may prove advantageous because of their more rapid washout and their minimal effect on hypoxic sensitivity at subanesthetic concentrations (Fig. 17-26). The effects of the volatile anesthetics on hypoxic drive may play an even more important role in patients who rely on hypoxic drive to set their level of ventilation, such as those with chronic respiratory failure or patients with obstructive sleep apnea.
Bronchiolar Smooth Muscle Tone
Bronchoconstriction under anesthesia occurs because of direct stimulation of the laryngeal and tracheal areas, from the administration of adjuvant drugs that cause histamine release, and from noxious stimuli activating vagal afferent nerves. The reflex response to these stimuli may be enhanced in lightly anesthetized patients.108 The responses also are enhanced in patients with known reactive airway disease, including those requiring bronchodilator therapy or those with chronic smoking histories. Airway smooth muscle extends as far distally as the terminal bronchioles and is under the influence of both parasympathetic and sympathetic nerves. The parasympathetic nerves mediate baseline airway tone and reflex bronchoconstriction via M2 and M3 muscarinic receptors on the airway smooth muscle, which initiate increases in intracellular cyclic guanosine monophosphate. Adrenergic receptors also are located on bronchial smooth muscle, and the β2-receptor subtype plays the predominant role in promoting bronchiolar muscle relaxation through an increase in intracellular cyclic adenosine monophosphate. The volatile anesthetics relax airway smooth muscle primarily by directly depressing smooth muscle contractility and indirectly inhibiting the reflex neural pathways.109 Direct effects of the volatile anesthetics partially depend on an intact bronchial epithelium, suggesting that epithelial damage or inflammation secondary to asthma may lessen their bronchodilating effect. The volatile anesthetics also may have protective effects by acting on the bronchial epithelium via a nonadrenergic, noncholinergic mechanism, possibly involving the nitric oxide pathway.110 Desflurane administration shortly after thiopental induction and tracheal intubation results in a transient increase in respiratory system resistance (bronchoconstriction), and this has been attributed to a direct effect of the pungency and airway irritability of desflurane (Fig. 17-27). This effect is worsened in patients with an active smoking history.111 Volatile anesthetics have been used effectively to treat status asthmaticus when other conventional treatments have failed.112,113 Although halothane has been historically used in these situations, it is no longer available commercially. Sevoflurane may be a better choice because of its quick onset, lack of pungency, lack of cardiovascular depression, and lower risk of cardiac arrhythmias compared with halothane.
P.435
|
|
|
Figure 17-26. Influence of 0.1 minimum alveolar concentration (MAC) of five volatile anesthetic agents on the ventilatory response to a step decrease in end-tidal oxygen concentration. Values are mean ± SD. Subanesthetic concentrations of the volatile anesthetics, except desflurane and sevoflurane, profoundly depress the response to hypoxia. (Adapted from Sarton E, Dahan A, Teppema L, et al: Acute pain and central nervous system arousal do not restore impaired hypoxic ventilatory responses during sevoflurane sedation. Anesthesiology 1996; 85: 295.) |
|
|
|
Figure 17-27. Changes in respiratory system resistance expressed as a percentage of the baseline recorded after tracheal intubation but prior to administration of sevoflurane or desflurane to the inspired gas mixture. Airway resistance responses to sevoflurane were significantly different from desflurane (*p < 0.05). (Adapted from Goff MJ, Arain SR, Ficke DJ, et al: Absence of bronchodilation during desflurane anesthesia: A comparison to sevoflurane and thiopental. Anesthesiology 2000; 93: 404.) |
Mucociliary Function
Ciliated respiratory epithelium extends from the trachea to the terminal bronchioles. Cells and glands in the tracheobronchial tree secrete mucus that captures surface particles for transport via ciliary action. There are a number of factors involved in diminished mucociliary function, particularly in the mechanically ventilated patient in whom dried, inspired gases impair ciliary movement, thicken the protective mucus, and reduce the ability of mucociliary function to transport surface particles out of the airway. Volatile anesthetics and nitrous oxide reduce ciliary movement and alter the characteristics of mucus. It also is known that smokers have impaired mucociliary function compared with nonsmokers, and the combination of a volatile anesthetic in a smoker who is mechanically ventilated sets up a scenario for inadequate clearing of secretions, mucus plugging, atelectasis, and hypoxemia.
Pulmonary Vascular Resistance
Although vascular smooth muscle is clearly affected by the volatile anesthetics, the pulmonary vascular relaxation from clinically relevant concentrations of inhaled anesthetics is minimal. In addition, an anesthetic-related decrease in cardiac output tends to offset the direct vasodilator action of the anesthetic, resulting in little or no change in pulmonary artery pressures and pulmonary blood flow. Even nitrous oxide, which has little effect on cardiac output and pulmonary blood flow, has at best a small effect to increase pulmonary vascular resistance. However, the effect of nitrous oxide may be magnified in patients with resting pulmonary hypertension.114
Perhaps more important in terms of volatile anesthetics and pulmonary blood flow is their potential to attenuate hypoxic pulmonary vasoconstriction (HPV). During periods of hypoxemia, HPV reduces blood flow to underventilated areas of the lung, thereby diverting blood flow to areas of the lung with greater ventilation. The net effect is to improve the V/Q matching, resulting in a reduced amount of venous admixture and improved arterial oxygenation. Although all of the inhaled anesthetics in high concentrations have been shown to attenuate HPV in animal models, the situation is less clear in patient studies. This may reflect the multifactorial effects of the volatile anesthetics on factors involved in pulmonary blood flow, including their cardiovascular, autonomic, and humoral actions. Furthermore, nonpharmacologic variables impair HPV, including surgical trauma, temperature, pH, Paco2, size of the hypoxic segment, and intensity of the hypoxic stimulus. One-lung ventilation (OLV) serves as a model where HPV should lessen the expected decrease in Pao2 and intrapulmonary shunt fraction (Qs/Qt). In patients undergoing OLV during thoracic surgery, volatile anesthetics have had minimal effects on Pao2 and Qs/Qt when changing from two-lung to OLV (Fig. 17-28).115,116 The efficacy of HPV to lessen shunt fraction varies inversely with pulmonary blood flow (and cardiac output). Isoflurane, sevoflurane, and desflurane preserve cardiac output and have minimal-to-modest effects on shunt fraction during OLV. Propofol appears to be no more beneficial on shunt fraction during OLV compared with sevoflurane.117
Hepatic Effects
Postoperative liver dysfunction, to varying degrees, has been associated with all of the volatile anesthetics in current use. There are two distinct mechanisms by which anesthetics have caused hepatitis. One is more common and related to hepatocyte toxicity. It is relatively mild, does not require a previous exposure, and has a low morbidity. The second is associated with repeat exposure and probably represents an immune reaction to oxidatively derived metabolites of anesthetics. It has been associated with severe liver damage and fulminant hepatic failure and is discussed later in the chapter.
|
|
|
Figure 17-28. Shunt fraction (top panel) and the alveolar-arterial oxygen gradient (bottom) immediately before, during, and after one-lung ventilation (OLV) in patients anesthetized with desflurane or isoflurane. Data are means. (Adapted from Pagel PS, Fu JL, Damask MC, et al: Desflurane and isoflurane produce similar alterations in systemic and pulmonary hemodynamics and arterial oxygenation in patients undergoing one-lung ventilation during thoracotomy. Anesth Analg 1998; 87: 800.) |
P.436
|
|
|
Figure 17-29. Changes (%, mean ± SE) in hepatic blood flow during administration of isoflurane or halothane. Decreases in portal vein blood flow produced by 2 minimum alveolar concentration (MAC) isoflurane are offset by increases in hepatic artery blood flow (autoregulation). Halothane resulted in decreases in both portal vein and hepatic artery blood flow, thereby significantly compromising total hepatic artery blood flow. (Adapted from Gelman S, Fowler KC, Smith LR: Liver circulation and function during isoflurane and halothane anesthesia. Anesthesiology 1984; 61: 726.) |
Hypoxic injury to hepatocytes can be a significant contributor to postoperative hepatic injury. The liver has two blood supplies. One is the well-oxygenated blood from the hepatic artery and the second is the poorly oxygenated blood from the portal vein. A positive attribute of the ether-based anesthetics (isoflurane, sevoflurane, and desflurane) is their ability to maintain or increase hepatic artery blood flow while decreasing (or not changing) portal vein blood flow (Fig. 17-29).118 Surgery in the area of the liver (or elsewhere in the abdominal cavity) that might compromise hepatic blood flow puts patients at risk for hepatic cell injury. In addition, hepatic enzyme induction, which increases oxygen demand, enhances the vulnerability of patients. Furthermore, patients who are critically dependent on oxygen supply for survival of remaining liver tissue, such as the cirrhotic patient, are at a higher risk for further hepatic injury than noncirrhotic individuals. Altered liver function tests have been used as an index of hepatic injury during anesthesia. Transient increases in plasma alanine aminotransferase activity do not occur in human volunteers administered desflurane, isoflurane, or sevoflurane,118,119,120 nor following sevoflurane or desflurane anesthesia in pediatric patients.121 Increases in the alanine aminotransferase or aspartate aminotransferase may not accurately reflect the extent of hepatic injury and are not uniquely specific to the liver. The centrilobular area of the liver is most susceptible to hypoxia. Therefore, a more sensitive measure of injury may be α-glutathione S-transferase (GST), since it is distributed primarily in the centrilobular hepatocytes. In patient studies, isoflurane did not increase GST.122 In elderly patients with no pre-existing liver disease and having peripheral surgery under sevoflurane or desflurane, a brief impairment of splanchnic blood flow was demonstrated and this led to increases in GST that resolved in 24 hours.123The possibility that the organ protection from sevoflurane would lessen hepatic disturbances after CABG surgery with cardiopulmonary bypass when compared with propofol was explored in a prospective randomized study of 320 patients. Postoperative biochemical markers of hepatic dysfunction were lower after the sevoflurane-based anesthetic.93 Thus, in terms of hepatocyte hypoxia, the modern-day volatile anesthetics have minimal adverse effects and might even afford protection from ischemic/hypoxic injury to cells.
Neuromuscular System and Malignant Hyperthermia
The inhaled anesthetics have two important actions on neuromuscular function. They directly relax skeletal muscle and they potentiate the action of neuromuscular blocking drugs.124,125 In contrast, nitrous oxide does not relax skeletal muscles. The direct effects of volatile anesthetics to relax skeletal muscle are most prominent above 1 MAC and can be further enhanced, by 40%, in patients with myasthenia gravis.126
Volatile anesthetic potentiation of neuromuscular blockade has been well documented. For example, the infusion rate of rocuronium required to maintain neuromuscular blockade is 30 to 40% less during isoflurane, desflurane, and sevoflurane compared with propofol.127 A similar left shift in the dose-response relationship has been observed with cisatracurium during volatile anesthetic administration versus during intravenous anesthesia.125 While the mechanism of volatile anesthetic potentiation of the neuromuscular blocking drugs is not entirely clear, it appears to be largely because of a postsynaptic effect at the nicotinic acetylcholine receptor located at the neuromuscular junction. Specifically, at the receptor level, the volatile anesthetics act synergistically with the neuromuscular blocking drugs to enhance their action.128 The degree of enhancement is related to their aqueous concentration so that at equi-MAC concentrations, the less potent anesthetics (e.g., desflurane and sevoflurane vs. isoflurane) should have a greater inhibitory effect on neuromuscular transmission. Support for this concept comes from a clinical study demonstrating 20% lower requirement for vecuronium to maintain a stable twitch depression during 1.25% desflurane compared with 1.25% isoflurane.129 However, desflurane and isoflurane (and sevoflurane) at equipotent concentrations acted similarly to enhance the effect of cisatracurium on neuromuscular function.125 This may relate to structural differences of the benzylisoquinolines versus aminosteroid neuromuscular blocking drugs.
All of the potent volatile anesthetics serve as triggers for malignant hyperthermia (MH) in genetically susceptible patients.130,131 In contrast, nitrous oxide is only a weak trigger for MH.132 The augmentation of caffeine-induced contractures by nitrous oxide is 1.3-fold, by isoflurane is 3-fold, and by halothane, 11-fold.132 Although desflurane is a weak trigger for MH, it has been associated with an unusual delayed onset of symptoms of MH in animals and humans.131,133
Genetic Effects, Obstetric use, and Effects on Fetal Development
The potential for genetic toxicity from volatile anesthetics seems minimal. The Ames test identifies chemicals that act as mutagens and carcinogens and has been negative for
P.437
isoflurane, desflurane, sevoflurane, and nitrous oxide.134,135 A possible genotoxic effect of desflurane has been detected in female patients with a cytogenetic assay that detects sister chromatid exchanges (SCE) in lymphocytes from peripheral blood. Desflurane transiently increased the frequency of SCE, whereas in children, sevoflurane did not increase SCE.136,137 The clinical implications of these findings are not clear in relation to the negative Ames test.
Volatile anesthetics can be teratogenic in animals,138 but none have been shown to be teratogenic in humans. Animal studies have indicated that nitrous oxide exposure in the early periods of gestation may result in adverse effects, including an increased incidence of fetal resorption.139 The same vulnerability does not exist during the administration of the potent volatile anesthetics.139
Nitrous oxide decreases the activity of vitamin B12-dependent enzymes, methionine synthetase and thymidylate synthetase. The mechanism appears to be an irreversible oxidation of the cobalt atom of vitamin B12 by nitrous oxide. When 70% nitrous oxide is administered to patients, the time to 50% inactivation of methionine synthetase is 46 minutes. The concern that these changes might have an effect on a rapidly developing embryo/fetus seems appropriate because methionine synthetase and thymidylate synthetase are involved in the formation of myelin and the formation of DNA, respectively. Inhibition of these enzymes could manifest as depression of bone marrow function and neurologic disturbances. In fact, megaloblastic changes in bone marrow are consistently observed in patients exposed to nitrous oxide for 24 hours, and 4 days of exposure to nitrous oxide has resulted in agranulocytosis. Furthermore, animals exposed to 15% nitrous oxide for several weeks developed neurologic changes including spinal cord and peripheral nerve degeneration and ataxia. A sensory motor polyneuropathy that is often combined with signs of posterior lateral spinal cord degeneration has been described in humans who chronically inhale nitrous oxide for recreational use.140 These effects have been attributed to reduced activity of the vitamin B12-dependent enzymes.
Uterine smooth muscle tone is diminished by volatile anesthetics in similar fashion to the effects of volatile anesthetics on vascular smooth muscle. There is a dose-dependent decrease in spontaneous myometrial contractility that is consistent among the volatile anesthetics.141,142 Desflurane and sevoflurane also inhibit the frequency and amplitude of myometrial contractions induced by oxytocin in a dose-dependent manner.141 Uterine relaxation/atony can become problematic at concentrations of volatile anesthesia >1 MAC, and might delay the onset time of newborn respiration.143 Consequently, a common technique used to provide general anesthesia for urgent cesarean sections is to administer low concentrations of the volatile anesthetic, such as 0.5 to 0.75 MAC, combined with nitrous oxide. This decreases the likelihood of uterine atony and blood loss, especially at a time after delivery when oxytocin responsiveness of the uterus is essential.143 In some situations, uterine relaxation may be desirable, such as to remove a retained placenta. In this case, a brief, high concentration of a volatile anesthetic may be advantageous.
There has been an ongoing concern about the incidence of spontaneous abortions in operating room personnel chronically exposed to trace concentrations of inhaled anesthetics, especially nitrous oxide.144 Early epidemiologic studies suggested that operating room personnel had an increased incidence of spontaneous abortions and congenital abnormalities in offspring. However, subsequent analysis of the data suggests inaccurate study design, confounding variables, and nonresponders might have led to flawed conclusions.145 In prospective studies, no causal relationship has been shown between exposure to waste anesthetic gases, regardless of the presence or absence of scavenging systems, and adverse health effects. Despite the unproven influence of trace concentrations of the volatile anesthetics on fetal development and spontaneous abortions, concerns for an adverse influence have resulted in the use of scavenging systems to remove anesthetic gases from the operating room and the establishment of standards for waste gas exposure. The National Institute for Occupational Safety and Health has recommended exposure levels for nitrous oxide is 25 parts per million (ppm) as a time-weighted average over 8 hours. The 1-hour exposure limit for halogenated anesthetics without nitrous oxide exposure is 2 ppm, and with nitrous oxide is 0.5 ppm.
In terms of neonatal effects from general anesthesia, Apgar scores and acid-base balance are not affected by anesthetic technique, such as spinal versus general.146 More sensitive measures of neurologic and behavioral function, such as the Scanlon Early Neonatal Neurobehavioral Scale and the Neurologic and Adaptive Capacities Score (NACS) indicate some transient depression of scores following general anesthesia that resolves at 24 hours after delivery.146,147
Anesthetic Degradation by Carbon Dioxide Absorbers
Compound A
Sevoflurane undergoes base-catalyzed degradation in carbon dioxide absorbents to form a vinyl ether called compound A. The production of compound A is enhanced in low flow or closed circuit breathing systems and by warm or very dry CO2 absorbents.148,149 Barium hydroxide lime produces more compound A than soda lime and this can be attributed to slightly higher absorbent temperature during CO2 extraction (Fig. 17-30).150 Desiccated barium hydroxide lime also has been implicated in the heat and fires associated with sevoflurane, discussed later. This absorbent has been removed from the U.S. market.
There are well-defined species differences in the threshold for compound A-induced nephrotoxicity. The threshold is approximately 300 ppm.hr in 250-g rats, >612 ppm.hr in pigs, and between 600 and 800 ppm.hr in monkeys. In patients and volunteers receiving sevoflurane in closed circuit or low flow delivery systems, inspired compound A concentrations averaged 8 to 24 and 20 to 32 ppm with soda lime and barium hydroxide lime, respectively.151,152,153,154 Total exposures as high as
P.438
320 to 400 ppm.hr have had no clear effect on clinical markers of renal function.155,156,157 In randomized and prospective volunteer and patient studies, no adverse renal effects from low-flow (0.5 to 1.0 L/min) or closed circuit sevoflurane anesthesia were detected using both standard clinical markers of renal function (serum creatinine and blood urea nitrogen concentrations) and experimental markers of renal function and structural integrity (proteinuria, glucosuria and enzymuria).152,153,154,158,159,160,161 In a prospective, multicenter, randomized study in patients with pre-existing renal disease, there were no adverse renal effects of long duration, low-flow sevoflurane.162,163 In fact, transient proteinuria, glucosuria, and enzymuria have been noted after desflurane, isoflurane, and propofol anesthesia indicating these elevated markers of renal injury might represent common effects unrelated to the choice of anesthetic.151,152 The majority of countries that have approved sevoflurane for clinical use have no flow restriction because of the proven safety of the anesthetic in scientific studies, where there has not been a single case report of renal injury from sevoflurane after a decade of use, despite a sensitized anesthesia community.
|
|
|
Figure 17-30. Compound A levels produced from three carbon dioxide absorbents during 1 minimum alveolar concentration sevo-flurane anesthesia delivered to volunteers at 1 L/min fresh gas flow (mean ± SE). Gas samples were taken from the inspired limb of the anesthesia circuit. *Different from barium hydroxide lime or soda lime (p <0.05). (Adapted from Mchaourab A, Arain SR, Ebert TJ: Lack of degradation of sevoflurane by a new carbon dioxide absorbent in humans. Anesthesiology 2001; 94: 1007.) |
Carbon Monoxide and Heat
CO2 absorbents degrade sevoflurane, desflurane, and isoflurane to carbon monoxide when the normal water content of the absorbent (13 to 15%) is markedly decreased <5%. 164,165,166The degradation is the result of an exothermic reaction of the anesthetics with the absorbent. The anesthetic molecular structure and the presence of a strong base in the carbon dioxide absorbent are involved in the formation of carbon monoxide (CO).165 Desflurane and isoflurane contain a difluoromethoxy moiety that is essential for the formation of CO. When studies are conducted with CO2 absorbents maintained at or just above room temperature, desflurane given at just under 1 MAC produced up to 8,000 ppm of CO versus 79 ppm with nearly 2 MAC sevoflurane.166 In desiccated barium hydroxide, CO production from desflurane was nearly threefold higher than with soda lime but was trivial with sevoflurane. In normal clinical use, CO2 canister temperatures are 25 to 45°C, but can be higher when employing a very low fresh gas flow. In a laboratory setting, when CO2 canister temperature is not controlled and sevoflurane is administered to desiccated barium hydroxide, the exothermic reaction can substantially increase canister temperatures. If the canister temperature exceeds 80°C, significant CO production is noted with sevoflurane.164 Instances of CO poisoning of patients have been reported in situations where the CO2 absorbent has been presumably dried (desiccated) because an anesthetic machine has been left on with a high fresh gas flow passing through the CO2 absorbent over an extended period of time.167,168,169,170 In an experimental setting, overnight drying of barium hydroxide for 14 hours at 10 L/min fresh gas flow did not result in significant CO production from desflurane, whereas 24 to 66 hours of fresh gas flow drying produced significant CO production.171
Although desflurane produces the most CO with desiccated CO2 absorbers, the reaction with sevoflurane produces the most heat.172 The strong exothermic reaction has caused significant heat production, fires, and patient injuries.173,174,175 Although sevoflurane is not flammable at <11%, formaldehyde, methanol, and formate have been identified,176 and these alone or in combination with oxygen might be flammable at high canister temperatures. In experimental settings, long exposure of 1 MAC sevoflurane to desiccated barium hydroxide resulted in canister temperatures in excess of 300°C, which can be associated with smoldering, melting of plastic components, explosions, and fires.164 Barium hydroxide has been removed from the U.S. market.
There are newer CO2 absorbents that do not degrade anesthetics (to either compound A or carbon monoxide), and they should reduce exothermic reactions. “From a patient safety perspective, widespread adoption of a nondestructive CO2 absorbent should be axiomatic.”177 Although the cost of these new CO2 absorbents Amsorb Plus (Armstrong Medical, Coleraine, UK) and DrägerSorb Free (Dräger, Lübek, Germany) is higher and the absorptive capacity may be lower than either barium hydroxide lime or soda lime, their benefit may be substantial. The use of a nondestructive absorbent eliminates all of the potential complications related to anesthetic breakdown and therefore minimizes the possibility of additional costs from those complications, including additional laboratory tests, hospital days, and medical/legal expenses. Adoption of these new absorbents into routine clinical practice is consistent with the patient safety goals of our anesthesia society.
Generic Sevoflurane Formulations
Generic formulations of sevoflurane were introduced into the clinical market in 2006. The methods for synthesizing sevoflurane differ between manufacturers.178 Although the active ingredient of sevoflurane from different manufacturers is chemically equivalent, the water content in the formulations differs and this accounts for their different resistance to degradation to hydrogen fluoride when exposed to a Lewis acid (metal halides and metal oxides that are present in modern-day vaporizers). Adding water to the formulation inhibits the action of Lewis acids to degrade sevoflurane to hydrogen fluoride. The formulation of Abbott Labs was changed to contain 300 to 400 ppm of water, based on an early adverse experience with hydrogen fluoride formation from a low water formulation in 1996. The generic formulation marketed by Baxter Laboratories is low in water (~65 ppm) and has been shown in clinical and laboratory studies to degrade to toxic and corrosive hydrogen fluoride.179 Recent reports indicate that the Penlon Sigma Delta sevoflurane vaporizer can degrade the Baxter low-water formulation of sevoflurane, resulting in etching of site glass and corrosion of the plastic on the vaporizer and discoloration of the anesthetic.180 Whether these differences in formulation lead to patient safety issues remains to be seen.
Anesthetic Metabolism
Fluoride-Induced Nephrotoxicity
The metabolism of enflurane may result in a well-described injury to renal collecting tubules.181,182 The nephrotoxicity presents as a high-output renal insufficiency that is unresponsive to vasopressin and is characterized by dilute polyuria, dehydration, serum hypernatremia, hyperosmolality, elevated blood urea nitrogen, and creatinine. An association between increased plasma fluoride concentrations and metabolism led to a “fluoride hypothesis.” This hypothesis has been re-examined recently in part because sevoflurane undergoes 5% metabolism that results in transient increases in serum fluoride concentrations, but it has not been associated with a renal-concentrating defect. The traditional hypothesis stated that both the duration of the high systemic fluoride concentrations (area under the fluoride-time curve) and the peak fluoride concentration (peaks above 50 µM appear to represent the toxic threshold) were related to nephrotoxicity (Fig. 17-31). The safety of sevoflurane with regard to fluoride concentrations may be the result of a rapid decline in plasma fluoride concentrations because of less availability of the anesthetic for metabolism from a faster washout compared with enflurane.183 In addition, the site of metabolism is an important
P.439
factor in toxicity, that is, intrarenal metabolism contributes to nephrotoxicity. Therefore, the potential for toxicity from relatively high plasma levels of fluoride following long exposure to sevoflurane is offset by the minimal amount of renal defluorination and this may explain its relative absence of renal-concentrating defects.184
|
|
|
Figure 17-31. Plasma inorganic fluoride concentrations (mean ± SE) before and after 2 to 4 hours of methoxyflurane, enflurane, sevoflurane, isoflurane, and desflurane anesthesia. (Adapted from Kharasch ED, Armstrong AS, Gunn K, et al: Clinical sevoflurane metabolism and disposition. II. The role of cytochrome P450 2E1 in fluoride and hexafluoroisopropanol formation. Anesthesiology 1995; 82: 1379; Mazze RI: Metabolism of the inhaled anaesthetics: Implications of enzyme induction. Br J Anaesth 1984; 56: 27S; and Sutton TS, Koblin DD, Gruenke LD, et al: Fluoride metabolites after prolonged exposure of volunteers and patients to desflurane. Anesth Analg 1991; 73: 180.) |
Factors such as total dose of anesthetic, liver enzyme induction, and obesity have been proven to enhance biotransformation. The activity of hepatic cytochrome P450 enzymes is increased by a variety of drugs, including phenobarbital, phenytoin, and isoniazid. Obesity causes increased metabolism (defluorination) of isoflurane.185 However, the effects of obesity on the defluorination of sevoflurane are less clear.186
Clinical Utility of Volatile Anesthetics
For Induction of Anesthesia
The appeal of mask induction in the adult population centers on the potential safety and utility of this technique.187,188,189,190 Spontaneous ventilation is preserved with a gas induction since patients essentially regulate their own depth of anesthesia (too much sevoflurane would suppress ventilation). The availability of sevoflurane, which is potent, poorly soluble, and nonpungent, and therefore can be inhaled easily, has generated renewed interest in this technique.
Clinical studies indicate that stage two excitation is avoided with high concentrations of sevoflurane. The typical time to loss of consciousness is 60 seconds when delivering 8% sevoflurane via the face mask. Sevoflurane also has been administered by mask as an approach to the difficult adult airway because it preserves spontaneous ventilation and does not cause salivation.191 The traditional “awake look” in the suspected difficult airway (where intravenous drugs are titrated to a level that allows direct laryngoscopy in the awake patient) has been modified to consist of spontaneous ventilation of high concentrations of sevoflurane until laryngoscopic evaluation is tolerated. Laryngeal mask placement can be successfully achieved 2 minutes after administering 7% sevoflurane via the face mask.189 The addition of nitrous oxide to the inspired gas mixture does not add significantly to the induction sequence. The gas induction technique is improved by pretreatment with benzodiazepines and worsened with opioid pretreatment because of apnea.188 Importantly, patient acceptance of this technique has been relatively high, exceeding 90%.187
There are a number of techniques to administer sevoflurane via face mask. These include priming the circuit (emptying the rebreathing bag, opening the “pop-off” valve, dialing the vaporizer to 8% while using a fresh gas flow of 8 L/min, and maintaining this for 60 seconds prior to applying the face mask to the patient), a single-breath induction from end-expiratory volume to maximum inspired volume, or simply breathing while the vaporizer is set to 8%. All seem to have the successful end result of loss of consciousness, generally within 1 minute.
For Maintenance of Anesthesia
The volatile anesthetics are clearly the most popular drug used to maintain anesthesia. They are easily administered via inhalation, they are readily titrated, they have a high safety ratio in terms of preventing recall, and the depth of anesthesia can be quickly adjusted in a predictable way while monitoring tissue levels via end-tidal concentrations. They are effective regardless of age or body habitus. They have some properties that prove beneficial in the operating room, including relaxation of skeletal muscle, preservation of cardiac output and cerebral blood flow, relatively predictable recovery profiles, and organ protection from ischemic injury. Some of the drawbacks to the use of the current volatile anesthetics are their absence of analgesic effects, their association with postoperative nausea and vomiting, and their potential for carbon monoxide poisoning and hepatitis.
Pharmacoeconomics and Value-Based Decisions
In the current environment of cost containment, clinicians are constantly being pressured to use less expensive drugs, including antiemetics, neuromuscular blocking drugs, and volatile anesthetics. Factors involved in the value-based decision include the efficacy of the drug, the side effects, its direct costs, and its indirect effects. In terms of efficacy, all of the volatile anesthetics are reasonably similar; that is, they can be used to establish a state of anesthesia for surgical interventions and can be easily reversed. A common side effect of the volatile anesthetics is nausea and vomiting. The need for rescue medications to treat nausea and vomiting after volatile anesthesia needs to be considered in any legitimate cost analysis. Direct costs are not simply the cost per milliliter of liquid or cost per bottle of anesthetic. Rather, they reflect the combination of the potency of the drug to establish a MAC level, the fresh gas flow, and the cost of the anesthetic. Sevoflurane and isoflurane are generic products, whereas desflurane is still under patent protection. At 1 L/min fresh gas flow, delivering 1 MAC,
P.440
desflurane, sevoflurane, and isoflurane cost about US $11.00 per hour, US $5.00 per hour, and US $2.00 per hour, respectively, depending on the local cost of drug acquisition. The indirect costs are probably the most difficult to pinpoint, but may be the most important when evaluating the cost of using the new volatile anesthetics. Examples of indirect costs include costs associated with operating room time, time in the postanesthesia care unit versus bypassing the postanesthesia care unit to a step-down unit, labor costs, and outcome-related costs, such as litigation to defend a bad outcome from an anesthetic drug.
|
|
|
Figure 17-32. The recovery times to orientation after anesthesia of varying durations. With the less soluble anesthetic sevoflurane, the time to orientation was independent of the anesthetic duration. In contrast, long anesthetic durations with isoflurane were associated with delayed times to orientation. (Adapted from Ebert TJ, Robinson BJ, Uhrich TD, et al: Recovery from sevoflurane anesthesia: A comparison to isoflurane and propofol anesthesia. Anesthesiology 1998; 89: 1524.) |
One of the arguments for using sevoflurane and desflurane has been their relative speed in terms of emergence from anesthesia. This argument has been tempered somewhat by the basic knowledge that titration of the volatile anesthetics can speed emergence times. Even the more soluble drug, isoflurane, can be titrated downward based on clinical experience or with the aid of processed EEG monitors, permitting fast wake-ups regardless of the choice of anesthetic agent. However, there is strong evidence to support the use of the less-soluble (but more expensive) drugs in the longest surgical cases (Fig. 17-32).192 In these cases the high direct cost of the anesthetic is balanced by the much improved recovery profile including a more rapid time to emergence and a more rapid discharge from the recovery room. Curiously, the discharge advantage with the low-soluble anesthetics has been difficult to show after shorter surgical procedures.
References
1. Kety SS: The physiological and physical factors governing the uptake of anesthetic gases by the body. Anesthesiology 1950; 11: 517
2. Eger EI: Anesthetic Uptake and Action. Baltimore, Williams & Wilkins, 1974
3. Ebert TJ, Robinson BJ, Uhrich TD, et al: Recovery from sevoflurane anesthesia: A comparison to isoflurane and propofol anesthesia. Anesthesiology 1998; 89: 1524
4. Hettrick DA, Pagel PS, Kersten JR, et al: Cardiovascular effects of xenon in isoflurane-anesthetized dogs with dilated cardiomyopathy. Anesthesiology 1998; 89: 1166
5. Nakata Y, Goto T, Morita S: Comparison of inhalation inductions with xenon and sevoflurane. Acta Anaesthesiol Scand 1997; 41: 1157
6. Kaplan R, Abramowitz MD, Epstein BS: Nitrous oxide and air-filled balloon-tipped catheters. Anesthesiology 1981; 55: 71
7. Stanley TH, Kawamura R, Graves C: Effects of nitrous oxide on volume and pressure of endotracheal tube cuffs. Anesthesiology 1974; 41: 256
8. Munson ES, Merrick HC: Effect of nitrous oxide on venous air embolism. Anesthesiology 1966; 27: 783
9. Waun JE, Sweitzer RS, Hamilton WK: Effect of nitrous oxide on middle ear mechanics and hearing acuity. Anesthesiology 1987; 28: 846
10. Tinker JH, Sharbrough FW, Michenfelder JD: Anterior shift of the dominant EEG rhythm during anesthesia in the Java monkey: Correlation with anesthetic potency. Anesthesiology 1977; 46: 252
11. Gross JB, Alexander CM. Awakening concentrations of isoflurane are not affected by analgesic doses of morphine. Anesth Analg 1988; 67: 27
12. Katoh T, Suguro Y, Kimura T, et al: Cerebral awakening concentration of sevoflurane and isoflurane predicted during slow and fast alveolar washout. Anesth Analg 1993; 77: 1012
13. Roizen MF, Horrigan RW, Frazer BM: Anesthetic doses blocking adrenergic (Stress) and cardiovascular responses to incision—MAC BAR. Anesthesiology 1981; 54: 390
14. Liem EB, Lin C-M, Suleman M-I, et al: Anesthetic requirement is increased in redheads. Anesthesiology 2004; 101: 279
15. Mogil JS, Wilson SG, Chesler EJ, et al: The nelanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc Natl Acad Sci U S A 2003; 100: 4867
16. Stekiel TA, Contney SJ, Bosnjak ZJ, et al: Reversal of minimum alveolar concentrations of volatile anesthetics by chromosomal substitution. Anesthesiology 2004; 101: 796
17. LeDez KM, Lerman J: The minimum alveolar concentration (MAC) of isoflurane in preterm neonates. Anesthesiology 1987; 67: 301
18. Mapleson WW: Effect of age on MAC in humans: a meta-analysis. Br J Anaesth 1996; 76: 179
19. Smith AL, Wollman H: Cerebral blood flow and metabolism: Effects of anesthetic drugs and techniques. Anesthesiology 1972; 36: 378
20. Scheller MS, Nakakimura K, Fleischer JE, et al: Cerebral effects of sevoflurane in the dog: Comparison with isoflurane and enflurane. Br J Anaesth 1990; 65: 388
21. Lutz LJ, Milde JH, Milde LN: The cerebral functional, metabolic, and hemodynamic effects of desflurane in dogs. Anesthesiology 1990; 73: 125
22. Fujibayashi T, Sugiura Y, Yanagimoto M, et al: Brain energy metabolism and blood flow during sevoflurane and halothane anesthesia: effects of hypocapnia and blood pressure fluctuations. Acta Anaesthesiol Scand 1994; 38: 413
23. Yli-Hankala A, Vakkuri A, Särkelä M, et al: Epileptiform electroencephalogram during mask induction of anesthesia with sevoflurane. Anesthesiology 1999; 91: 1596
24. Jääskeläinen SK, Kaisti K, Suni L, et al: Sevoflurane is epileptogenic in healthy subjects at surgical levels of anesthesia. Neurology 2003; 61: 1073
25. Julliac B, Guehl D, Chopin F, et al: Sharp increase in cerebral sevoflurane concentration during mask induction in adults is a major risk factor of spike wave occurrence. Anesthesiology, 2004: A-132
26. Hisada K, Morioka T, Fukui K, et al: Effects of sevoflurane and isoflurane on electrocorticographic activities in patients with temporal lobe epilepsy. J Neurosurg Anesthesiol 2001; 13: 333
27. Algotsson L, Messeter K, Nordström CH, et al: Cerebral blood flow and oxygen consumption during isoflurane and halothane anesthesia in man. Acta Anaesthesiol Scand 1988; 32: 15
28. Lutz LJ, Milde JH, Milde LN: The response of the canine cerebral circulation to hyperventilation during anesthesia with desflurane. Anesthesiology 1991; 74: 504
29. Adams RW, Cucchiara RF, Gronert GA, et al: Isoflurane and cerebrospinal fluid pressure in neurosurgical patients. Anesthesiology 1981; 54: 97
30. Grosslight K, Foster R, Colohan AR, et al: Isoflurane for neuroanesthesia: risk factors for increases in intracranial pressure. Anesthesiology 1985; 63: 533
31. Artru AA: Isoflurane does not increase the rate of CSF production in the dog. Anesthesiology 1984; 60: 193
32. Talke P, Caldwell JE, Richardson CA: Sevoflurane increases lumbar cerebrospinal fluid pressure in normocapnic patients undergoing transsphenoidal hypophysectomy. Anesthesiology 1999; 91: 127
33. Talke P, Caldwell J, Dodsont B, et al: Desflurane and isoflurane increases lumbar cerebrospinal fluid pressure in normocapnic patients undergoing transsphenoidal hypophysectomy. Anesthesiology 1996; 85: 999
34. Michenfelder JD, Milde JH, Sundt JM Jr: Cerebral protection by barbiturate anesthesia. Use after middle cerebral artery occlusion in Java monkeys. Arch Neurol 1976; 33
35. Muzzi DA, Losasso TJ, Dietz NM, et al: The effect of desflurane and isoflurane on cerebrospinal fluid pressure in humans with supratentorial mass lesions. Anesthesiology 1992; 76: 720
36. Sponheim S, Skraastad Ø, Helseth E, et al: Effects of 0.5 and 1.0 MAC isoflurane, sevoflurane and desflurane on intracranial and cerebral perfusion pressures in children. Acta Anaesthesiol Scand 2003; 47: 932
37. Sugioka S: Effects of sevoflurane on intracranial pressure and formation and absorption of cerebrospinal fluid in cats. [Japanese]. Masui. Jpn J Anesthesiol 1992; 41: 1434
38. Artru AA: Rate of cerebrospinal fluid formation, resistance to reabsorption of cerebrospinal fluid, brain tissue water content, and electroencephalogram during desflurane anesthesia in dogs. J Neurosurg Anesthesiol 1993; 5: 178
P.441
39. Bundgaard H, von Oettingen G, Larsen KM, et al: Effects of sevoflurane on intracranial pressure, cerebral blood flow and cerebral metabolism. Acta Anaesthesiol Scand 1998; 42: 621
40. Seyde WC, Longnecker DE: Cerebral oxygen tension in rats during deliberate hypotension with sodium nitroprusside, 2-chloroadenosine, or deep isoflurane anesthesia. Anesthesiology 1986; 64: 480
41. Engelhard K, Werner C, Reeker W, et al: Desflurane and isoflurane improve neurological outcome after incomplete cerebral ischaemia in rats. Br J Anaesth 1999; 83: 415
42. Werner C, Möllenberg O, Kochs E, et al: Sevoflurane improves neurological outcome after incomplete cerebral ischaemia in rats. Br J Anaesth 1995; 75: 756
43. Loepke AW, Priestley MA, Schultz SEM, J. et al: Desflurane improves neurologic outcome after low-flow cardiopulmonary bypass in newborn pigs. Anesthesiology 2002; 97: 1521
44. Hoffman WE, Charbel FT, Edelman G, et al: Thiopental and desflurane treatment for brain protection. Neurosurgery 1998; 43: 1050
45. Sakabe T, Tsutsui T, Maekawa T, et al: Local cerebral glucose utilization during nitrous oxide and pentobarbital anesthesia in rats. Anesthesiology 1985; 63: 262
46. Fleischer JE, Milde JH, Moyer TP, et al: Cerebral effects of high-dose midazolam and subsequent reversal with Ro 15-1788 in dogs. Anesthesiology 1988; 68: 234
47. Hoffman WE, Miletich DJ, Albrecht RF: The effects of midazolam on cerebral blood flow and oxygen consumption and its interaction with nitrous oxide. Anesth Analg 1986; 65: 729
48. Hartung J, Cottrell JE: Nitrous oxide reduces thiopental-induced prolongation of survival in hypoxic and anoxic mice. Anesth Analg 1987; 66
49. Ebert TJ, Kampine JP: Nitrous oxide augments sympathetic outflow: Direct evidence from human peroneal nerve recordings. Anesth Analg 1989; 69: 444
50. Ebert TJ, Muzi M, Lopatka CW: Neurocirculatory responses to sevoflurane in humans. A comparison to desflurane. Anesthesiology 1995; 83: 88
51. Malan TP Jr, DiNardo JA, Isner RJ, et al: Cardiovascular effects of sevoflurane compared with those of isoflurane in volunteers. Anesthesiology 1995; 83: 918
52. Weiskopf RB, Cahalan MK, Eger EI II, et al: Cardiovascular actions of desflurane in normocarbic volunteers. Anesth Analg 1991; 73: 143
53. Stevens WC, Cromwell TH, Halsey MJ, et al: The cardiovascular effects of a new inhalation anesthetic, Forane, in human volunteers at a constant arterial carbon dioxide tension. Anesthesiology 1971; 35: 8
54. Calverley RK, Smith NT, Prys-Roberts C, et al: Cardiovascular effects of enflurane anesthesia during controlled ventilation in man. Anesth Analg 1978; 57: 619
55. Ebert TJ, Muzi M: Sympathetic hyperactivity during desflurane anesthesia in healthy volunteers. A comparison with isoflurane. Anesthesiology 1993; 79: 444
56. Weiskopf RB, Moore MA, Eger EI II, et al: Rapid increase in desflurane concentration is associated with greater transient cardiovascular stimulation than with rapid increase in isoflurane concentration in humans. Anesthesiology 1994; 80: 1035
57. Muzi M, Ebert TJ, Hope WG, et al: Site(s) mediating sympathetic activation with desflurane. Anesthesiology 1996; 85: 737
58. Weiskopf RB, Eger EI II, Noorani M, et al: Fentanyl, esmolol, and clonidine blunt the transient cardiovascular stimulation induced by desflurane in humans. Anesthesiology 1994; 81: 1350
59. Yonker-Sell AE, Muzi M, Hope WG, et al: Alfentanil modifies the neurocirculatory responses to desflurane. Anesth Analg 1996; 82: 162
60. Pacentine GG, Muzi M, Ebert TJ: Effects of fentanyl on sympathetic activation associated with the administration of desflurane. Anesthesiology 1995; 82: 823
61. Calverley RK, Smith NT, Jones CW, et al: Ventilatory and cardiovascular effects of enflurane anesthesia during spontaneous ventilation in man. Anesth Analg 1978 57: 610
62. Weiskopf RB, Cahalan MK, Ionescu P, et al: Cardiovascular actions of desflurane with and without nitrous oxide during spontaneous ventilation in humans. Anesth Analg 1991; 73: 165
63. Weiskopf RB, Holmes MA, Rampil IJ, et al: Cardiovascular safety and actions of high concentrations of I-653 and isoflurane in swine. Anesthesiology 1989; 70: 793
64. Ebert TJ: Differential effects of nitrous oxide on baroreflex control of heart rate and peripheral sympathetic nerve activity in humans. Anesthesiology 1990; 72: 16
65. Cahalan MK, Weiskopf RB, Eger EI II, et al: Hemodynamic effects of desflurane/nitrous oxide anesthesia in volunteers. Anesth Analg 1991; 73: 157
66. Theye RA, Michenfelder JD. Whole-body and organ VO2 changes with enflurane, isoflurane, and halothane. Br J Anaesth 1975; 47: 813
67. Crawford MW, Lerman J, Saldivia V, et al: Hemodynamic and organ blood flow responses to halothane and sevoflurane anesthesia during spontaneous ventilation. Anesth Analg 1992; 75: 1000
68. Ebert TJ, Harkin CP, Muzi M: Cardiovascular responses to sevoflurane: A review. Anesth Analg 1995; 81: S11
69. Atlee JL, III, Bosnjak ZJ: Mechanisms for cardiac dysrhythmias during anesthesia. Anesthesiology 1990; 72: 347
70. Bosnjak ZJ, Kampine JP: Effects of halothane, enflurane, and isoflurane on the SA node. Anesthesiology 1983; 58: 314
71. Hartman JC, Pagel PS, Kampine JP, et al: Influence of desflurane on regional distribution of coronary blood flow in a chronically instrumented canine model of multivessel coronary artery obstruction. Anesth Analg 1991; 72: 289
72. Hartman JC, Kampine JP, Schmeling WT, et al: Steal-prone coronary circulation in chronically instrumented dogs: isoflurane versus adenosine. Anesthesiology 1991; 74: 744
73. Kersten JR, Brayer AP, Pagel PS, et al: Perfusion of ischemic myocardium during anesthesia with sevoflurane. Anesthesiology 1994; 81: 995
74. Harkin CP, Pagel PS, Kersten JR, et al: Direct negative inotropic and lusitropic effects of sevoflurane. Anesthesiology 1994; 81: 156
75. Slogoff S, Keats AS, Dear WE, et al: Steal-prone coronary anatomy and myocardial ischemia associated with four primary anesthetic agents in humans. Anesth Analg 1991; 72: 22
76. O'Young J, Mastrocostopoulos G, Hilgenberg A, et al: Myocardial circulatory and metabolic effects of isoflurane and sufentanil during coronary artery surgery. Anesthesiology 1987; 66: 653
77. Tuman KJ, McCarthy RJ, Spiess BD, et al: Does choice of anesthetic agent significantly affect outcome after coronary artery surgery? Anesthesiology 1989; 70: 189
78. Ebert TJ, Kharasch ED, Rooke GA, et al: Myocardial ischemia and adverse cardiac outcomes in cardiac patients undergoing noncardiac surgery with sevoflurane and isoflurane. Anesth Analg 1997; 85: 993
79. Searle NR, Martineau RJ, Conzen P, et al: Comparison of sevoflurane/fentanyl and isoflurane/fentanyl during elective coronary artery bypass surgery. Can J Anaesth 1996; 43: 890
80. Thomson IR, Bowering JB, Hudson RJ, et al: A comparison of desflurane and isoflurane in patients undergoing coronary artery surgery. Anesthesiology 1991; 75: 776
81. Helman JD, Leung JM, Bellows WH, et al: The risk of myocardial ischemia in patients receiving desflurane versus sufentanil anesthesia for coronary artery bypass graft surgery. Anesthesiology 1992; 77: 47
82. Riess ML, Stowe DF, Warltier DC: Cardiac pharmacolocial preconditioning with volatile anesthetics: From bench to bedside? Am J Physiol 2004; 286: H1603
83. Stowe DF, Kevin LG: Cardiac preconditioning by volatile anesthetic agents: A defining role for altered mitochondrial bioenergetics. Antioxidants & Redox Signaling 2004; 6: 439
84. Novalija E, Kevin LG, Camara AK, et al: Reactive oxygen species precede the epsilon isoform of protein kinase C in the anesthetic preconditioning signaling cascade. Anesthesiology 2003; 99: 421
85. Kwok WM, Martinelli AT, Fujimoto K, et al: Differential modulation of the cardiac adenosine triphosphate-sensitive potassium channel by isoflurane and halothane. Anesthesiology 2002; 97: 50
86. Kevin LG, Katz P, Camara AK, et al: Anesthetic preconditioning: effects on latency to ischemic injury in isolated hearts. Anesthesiology 2003; 99: 385
87. De Hert SG, Turani F, Mathur S, et al: Cardioprotection with volatile anesthetics: mechanisms and clinical implications. Anesth Analg 2005; 100: 1584
88. Yu CH, Beattie WS: The effects of volatile anesthetics on cardiac ischemic complications and mortality in CABG: a meta-analysis. Can J Anaesth 2006; 53: 906
89. De Hert SG, Van der Linden PJ, Cromheecke S, et al: Choice of primary anesthetic regimen can influence intensive care unit length of stay after coronary surgery with cardiopulmonary bypass. Anesthesiology 2004; 101: 9
90. Gu W, Pagel PS, Warltier DC, et al: Modifying cardiovascular risk in diabetes mellitus. Anesthesiology 2003; 98: 774
91. Clarkson AN: Anesthetic-mediated protection/preconditioning during cerebral ischemia. Life Sci 2007; 80: 1157
92. Lee HT, Ota-Setlik A, Fu Y, et al: Differential protective effects of volatile anesthetics against renal ischemia-reperfusion injury in vivo. Anesthesiology 2004; 101: 1313
93. Lorsomradee S, Cromheecke S, Lorsomradee S, et al: Effects of sevoflurane on biomechanical markers of hepatic and renal dysfunction after coronary artery surgery. J Cardiothorac Vasc Anesth 2006; 20: 684
94. Seagard JL, Hopp FA, Bosnjak ZJ, et al: Sympathetic efferent nerve activity in conscious and isoflurane-anesthetized dogs. Anesthesiology 1984; 61: 266
95. Seagard JL, Hopp FA, Donegan JH, et al: Halothane and the carotid sinus reflex: evidence for multiple sites of action. Anesthesiology 1982; 57: 191
96. Seagard JL, Elegbe EO, Hopp FA, et al: Effects of isoflurane on the baroreceptor reflex. Anesthesiology 1983; 59: 511
97. Muzi M, Ebert TJ: A randomized, prospective comparison of halothane, isoflurane and enflurane on baroreflex control of heart rate in humans, Advances in Pharmacology, Vol. 31: Anesthesia and Cardiovascular Disease. Edited by Bosnjak Z, Kampine JP. San Diego, Academic Press, 1994, pp. 379
98. Ebert TJ, Perez F, Uhrich TD, et al: Desflurane-mediated sympathetic activation occurs in humans despite preventing hypotension and baroreceptor unloading. Anesthesiology 1998; 88: 1227
99. Muzi M, Ebert TJ: A comparison of baroreflex sensitivity during isoflurane and desflurane anesthesia in humans. Anesthesiology 1995; 82: 919
100. Tanaka M, Nishikawa T: Arterial baroreflex function in humans anaesthetized with sevoflurane. Br J Anaesth 1999; 82: 350
101. Ebert TJ, Kotrly KJ, Madsen KS, et al: Fentanyl-diazepam anesthesia with or without N2O does not attenuate cardiopulmonary baroreflex-mediated vasoconstrictor responses to controlled hypovolemia in humans. Anesth Analg 1988; 67: 548
P.442
102. Kotrly KJ, Ebert TJ, Vucins EJ, et al: Effects of fentanyl-diazepam-nitrous oxide anaesthesia on arterial baroreflex control of heart rate in man. Br J Anaesth 1986; 58: 406
103. Tanaka M, Nishikawa T: Sevoflurane speeds recovery of baroreflex control of heart rate after minor surgical procedures compared with isoflurane. Anesth Analg 1999; 89: 284
104. Muzi M, Lopatka CW, Ebert TJ: Desflurane-mediated neurocirculatory activation in humans: Effects of concentration and rate of change on responses. Anesthesiology 1996; 84: 1035
105. Calverley RK, Smith NT, Jones CW, et al: Ventilatory and cardiovascular effects of enflurane anesthesia during spontaneous ventilation in man. Anesth Analg 1978; 57: 610
106. Lopez-Barneo J, Pardal R, Ortega-Saenz P: Cellular mechanisms of oxygen sensing. Ann Rev Physiol 2001; 63: 259
107. Dahan A, Teppema LJ: Influence of anaesthesia and analgesia on the control of breathing. Br J Anaesth 2003; 91: 40
108. Hirshman CA, Bergman NA: Factors influencing intrapulmonary airway calibre during anaesthesia. Br J Anaesth 1990; 65: 30
109. Hirshman CA, Edelstein G, Peetz S, et al: Mechanism of action of inhalational anesthesia on airways. Anesthesiology 1982; 56: 107
110. Lindeman KS, Baker SG, Hirshman CA: Interaction between halothane and the nonadrenergic, noncholinergic inhibitory system in porcine trachealis muscle. Anesthesiology 1994; 81: 641
111. Goff MJ, Arain SR, Ficke DJ, et al: Absence of bronchodilation during desflurane anesthesia: A comparison to sevoflurane and thiopental. Anesthesiology 2000; 93: 404
112. Mori N, Nagata H, Ohta S, et al: Prolonged sevoflurane inhalation was not nephrotoxic in two patients with refractory status asthmaticus. Anesth Analg 1996; 83: 189
113. Johnston RG, Noseworthy TW, Friesen EG, et al: Isoflurane therapy for status asthmaticus in children and adults. Chest 1990; 97: 698
114. Reiz S: Nitrous oxide augments the systemic and coronary haemodynamic effects of isoflurane in patients with ischaemic heart disease. Acta Anaesthesiol Scand 1983; 27: 464
115. Benumof JL, Augustine SD, Gibbons JA: Halothane and isoflurane only slightly impair arterial oxygenation during one-lung ventilation in patients undergoing thoracotomy. Anesthesiology 1987; 67: 910
116. Pagel PS, Fu JL, Damask MC, et al: Desflurane and isoflurane produce similar alterations in systemic and pulmonary hemodynamics and arterial oxygenation in patients undergoing one-lung ventilation during thoracotomy. Anesth Analg 1998; 87: 800
117. Beck DH, Doepfmer UR, Sinemus C, et al: Effects of sevoflurane and propofol on pulmonary shunt fraction during one-lung ventilation for thoracic surgery. Br J Anaesth 2001; 86: 38
118. Frink EJ Jr, Ghantous H, Malan TP, et al: Plasma inorganic fluoride with sevoflurane anesthesia: Correlation with indices of hepatic and renal function. Anesth Analg 1992; 74: 231
119. Eger EI II: Isoflurane (Forane): A compendium and reference. 2nd ed. Madison, Ohio Medical Products, 1985
120. Weiskopf RB, Eger EI II, Ionescu P, et al: Desflurane does not produce hepatic or renal injury in human volunteers. Anesth Analg 1992; 74: 570
121. Isik Y, Goksu S, Kocoglu H, et al: Low flow desflurane and sevoflurane anaesthesia in children. Eur J Anaesthesiol 2006; 23: 60
122. Hussey AJ, Aldridge LM, Paul D, et al: Plasma glutathione-S-transferase concentration as a measure of hepatocellular integrity following a single general anaesthetic with halothane, enflurane or isoflurane. Br J Anaesth 1988; 60: 130
123. Suttner SW, Schmidt CC, Boldt J, et al: Low-flow desflurane and sevoflurane anesthesia minimally affect hepatic integrity and function in elderly patients. Anesth Analg 2000; 91: 206
124. Kurahashi K, Maruta H: The effect of sevoflurane and isoflurane on the neuromuscular block produced by vecuronium continuous infusion. Anesth Analg 1996; 82: 942
125. Wulf H, Kahl M, Ledowski T: Augmentation of the neuromuscular blocking effects of cisatracurium during desflurane, sevoflurane, isoflurane or total i.v. anaesthesia. Br J Anaesth 1998; 80: 308
126. Nitahara K, Sugi Y, Higa K, et al: Neuromuscular effects of sevoflurane in myasthenia gravis patients. Br J Anaesth 2007; 98: 337
127. Bock M, Klippel K, Nitsche B, et al: Rocuronium potency and recovery characteristics during steady-state desflurane, sevoflurane, isoflurane or propofol anaesthesia. Br J Anaesth 2000; 84: 43
128. Paul M, Fokt RM, Kindler CH, et al: Characterization of the interactions between volatile anesthetics and neuromuscular blockers at the muscle nicotinic acetylcholine receptor. Anesth Analg 2002; 95: 362
129. Wright PMC, Hart P, Lau M, et al: The magnitude and time course of vecuronium potentiation by desflurane versus isoflurane. Anesthesiology 1995; 82: 404
130. Ducart A, Adnet P, Renaud B, et al: Malignant hyperthermia during sevoflurane administration. Anesth Analg 1995; 80: 609
131. Allen GC, Brubaker CL: Human malignant hyperthermia associated with desflurane anesthesia. Anesth Analg 1998; 86: 1328
132. Reed SB, Strobel GE: An in vitro model of malignant hyperthermia: differential effects of inhalation anesthetics on caffeine-induced muscle contractures. Anesthesiology 1978; 48: 254
133. Papadimos TJ, Almasri M, Padgett JS, et al: A suspected case of delayed onset malignant hyperthermia with desflurane anesthesia. Anesth Analg 2004; 98: 548
134. Hobbhahn J, Wiesner G, Taeger K: [Occupational exposure and environmental pollution: the role of inhalation anesthetics with special consideration of sevoflurane.]. Anaesthesist. 1998; 47: S77
135. Baden J, Kelley M, Mazze R: Mutagenicity of experimental inhalational anesthetic agents: sevoflurane, synthane, diozychlorane, and dioxyflurane. Anesthesiology 1982; 56: 462
136. Krause T, Scholz J, Jansen L, et al: Sevoflurane anaesthesia does not induce the formation of sister chromatid exchanges in peripheral blood lymphocytes of children. Br J Anaesth 2003; 90: 233
137. Akin A, Ugur F, Ozkul Y, et al: Desflurane anaesthesia increases sister chromatid exchanges in human lymphocytes. Acta Anaesthesiol Scand 2005; 49: 1559
138. Mazze RI, Wilson AI, Rice SA, et al: Fetal development in mice exposed to isoflurane. Teratology 1985; 32: 339
139. Mazze RI, Fujinaga M, Rice SA, et al: Reproductive and teratogenic effects of nitrous oxide, halothane, isoflurane, and enflurane in Sprague-Dawley rats. Anesthesiology 1986; 64: 339
140. Layzer RB, Fishman RA, Schafer JA: Neuropathy following use of nitrous oxide. Neurology 1978; 28: 504
141. Yildiz K, Dogru K, Dalgic H, et al: Inhibitory effects of desflurane and sevoflurane on oxytocin-induced contractions of isolated pregnant human myometrium. Acta Anaesthesiol Scand 2005; 49: 1355
142. Munson ES, Embro WJ: Enflurane, isoflurane and halothane and isolated human uterine muscle. Anesthesiology 1977; 46: 11
143. Abboud TK, Zhu J, Richardson M, et al: Desflurane: a new volatile anesthetic for cesarean section. Maternal and neonatal effects. Acta Anaesthesiol Scand 1995; 39: 723
144. Lane GA, Nahrwold ML, Tait AR: Anesthetics as teratogens: Nitrous oxide is fetotoxic, xenon is not. Science 1980; 210: 899
145. McGregor DG: Occupational exposure to trace concentrations of waste anesthetic gases. Mayo Clin Proc 2000; 75: 273
146. Abboud TK, Nagappala S, Murakawa K, et al: Comparison of the effects of general and regional anesthesia for cesarean section on neonatal neurologic and adaptive capacity scores. Anesth Analg 1985; 64: 996
147. Warren TM, Datta S, Ostheimer GW, et al: Comparisons of the maternal and neonatal effects of halothane, enflurane and isoflurane for cesarean delivery. Anesth Analg 1983; 62: 516
148. Ruzicka JA, Hidalgo JC, Tinker JH, et al: Inhibition of volatile sevoflurane degradation product formation in an anesthesia circuit by a reduction in soda lime temperature. Anesthesiology 1994; 81: 238
149. Fang ZX, Kandel L, Laster MJ, et al: Factors affecting production of compound A from the interaction of sevoflurane with Baralyme and soda lime. Anesth Analg 1996; 82: 775
150. Frink EJ Jr, Malan TP, Morgan SE, et al: Quantification of the degradation products of sevoflurane in two CO2 absorbents during low-flow anesthesia in surgical patients. Anesthesiology 1992; 77: 1064
151. Ebert TJ, Arain SR: Renal effects of low-flow anesthesia with desflurane and sevoflurane in patients. Anesthesiology, 1999: A404
152. Kharasch ED, Frink EJ Jr, Zager R, et al: Assessment of low-flow sevoflurane and isoflurane effects on renal function using sensitive markers of tubular toxicity. Anesthesiology 1997; 86: 1238
153. Bito H, Ikeuchi Y, Ikeda K: Effects of low-flow sevoflurane anesthesia on renal function. Comparison with high-flow sevoflurane anesthesia and low-flow isoflurane anesthesia. Anesthesiology 1997; 86: 1231
154. Bito H, Ikeda K: Closed-circuit anesthesia with sevoflurane in humans. Effects on renal and hepatic function and concentrations of breakdown products with soda lime in the circuit. Anesthesiology 1994; 80: 71
155. Eger EI II, Koblin DD, Bowland T, et al: Nephrotoxicity of sevoflurane versus desflurane anesthesia in volunteers. Anesth Analg 1997; 84: 160
156. Ebert TJ, Frink EJ Jr, Kharasch ED: Absence of biochemical evidence for renal and hepatic dysfunction after 8 hours of 1.25 minimum alveolar concentration sevoflurane anesthesia in volunteers. Anesthesiology 1998; 88: 601
157. Eger EI II, Gong D, Koblin DD, et al: Dose-related biochemical markers of renal injury after sevoflurane versus desflurane anesthesia in volunteers. Anesth Analg 1997; 85: 1154
158. Groudine SB, Fragen RJ, Kharasch ED, et al: Comparison of renal function following anesthesia with low-flow sevoflurane and isoflurane. J Clin Anesth 1999; 11: 201
159. Bito H, Ikeda K: Renal and hepatic function in surgical patients after low-flow sevoflurane or isoflurane anesthesia. Anesth Analg 1996; 82: 173
160. Ebert TJ, Messana LD, Uhrich TD, et al: Absence of renal and hepatic toxicity after four hours of 1.25 minimum alveolar concentration sevoflurane anesthesia in volunteers. Anesth Analg 1998; 86: 662
161. Ebert TJ, Frink EJ Jr, Kharasch ED: Absence of biochemical evidence for renal and hepatic dysfunction after 8 hours of 1.25 minimum alveolar concentration sevoflurane anesthesia in volunteers. Anesthesiology 1998; 88: 601
162. Conzen PF, Kharasch ED, Czerner SFA, et al: Low-flow sevoflurane compared with low-flow isoflurane anesthesia in patients with stable renal insufficiency. Anesthesiology 2002; 97: 578
163. Litz RJ, Hübler M, Lorenz W, et al: Renal responses to desflurane and isoflurane in patients with renal insufficiency. Anesthesiology 2002; 97: 1133
P.443
164. Holak EJ, Mei DA, Dunning MB, III, et al: Carbon monoxide production from sevoflurane breakdown: Modeling of exposures under clinical conditions. Anesth Analg 2003; 96: 757
165. Baxter PJ, Garton K, Kharasch ED: Mechanistic aspects of carbon monoxide formation from volatile anesthetics. Anesthesiology 1998; 89: 929
166. Fang ZX, Eger EI II, Laster MJ, et al: Carbon monoxide production from degradation of desflurane, enflurane, isoflurane, halothane, and sevoflurane by soda lime and baralyme. Anesth Analg 1995; 80: 1187
167. Berry PD, Sessler DI, Larson MD: Severe carbon monoxide poisoning during desflurane anesthesia. Anesthesiology 1999; 90: 613
168. Woehlck HJ: Severe intraoperative CO poisoning. Anesthesiology 1999; 90: 353
169. Woehlck HJ, Dunning M, III, Gandhi S, et al: Indirect detection of intraoperative carbon monoxide exposure by mass spectrometry during isoflurane anesthesia. Anesthesiology 1995; 83: 213
170. Woehlck HJ, Dunning M, Connolly LA: Reduction in the incidence of carbon monoxide exposures in humans undergoing general anesthesia. Anesthesiology 1997; 87: 228
171. Woehlck HJ, Dunning M, III, Raza T, et al: Physical factors affecting the production of carbon monoxide from anesthetic breakdown. Anesthesiology 2001; 94: 453
172. Wissing H, Kuhn I, Warnken U, et al: Carbon monoxide production from desflurane, enflurane, halothane, isoflurane and sevoflurane with dry soda lime. Anesthesiology 2001; 95: 1205
173. Castro BA, Freedman LA, Craig WL, et al: Explosion within an anesthesia machine: Baralyme®, high fresh gas flows and sevoflurane concentration. Anesthesiology 2004; 101: 537
174. Wu J, Previte JP, Adler E, et al: Spontaneous ignition, explosion, and fire with sevoflurane and barium hydroxide lime. Anesthesiology 2004; 101: 534
175. Fatheree RS, Leighton BL: Acute respiratory distress syndrome after an exothermic Baralyme®-sevoflurane reaction. Anesthesiology 2004; 101: 531
176. Hanaki C, Fujii K, Morio M, et al: Decomposition of sevoflurane by sodalime. Hiroshima J Med Sci 1987; 36: 61
177. Kharasch ED: Putting the brakes on anesthetic breakdown. Anesthesiology 1999; 91: 1192
178. Baker MT: Sevoflurane: are there differences in products? Anesth Analg 2007; 104: 1447
179. Kharasch ED, Subbarao GN, Stephens DA, et al: Influence of sevoflurane formulation water content on degradation to hydrogen fluoride in vaporizers. Anesthesiology 2007; 107: A1591
180. O'Neill B, Hafiz MA, De Beer DA: Corrosion of Penlon sevoflurane vaporisers. Anaesthesia 2007; 62: 421
181. Frink EJ Jr, Malan TP Jr, Isner RJ, et al: Renal concentrating function with prolonged sevoflurane or enflurane anesthesia in volunteers. Anesthesiology 1994; 80: 1019
182. Mazze RI, Calverley RK, Smith NT: Inorganic fluoride nephrotoxicity: Prolonged enflurane and halothane anesthesia in volunteers. Anesthesiology 1977; 46: 265
183. Mazze RI: The safety of sevoflurane in humans. Anesthesiology 1992; 77: 1062
184. Kharasch ED, Hankins DC, Thummel KE: Human kidney methoxyflurane and sevoflurane metabolism. Intrarenal fluoride production as a possible mechanism of methoxyflurane nephrotoxicity. Anesthesiology 1995; 82: 689
185. Strube PJ, Hulands GH, Halsey MJ: Serum fluoride levels in morbidly obese patients: enflurane compared with isoflurane anaesthesia. Anaesthesia 1987; 42: 685
186. Frink EJ Jr, Malan TP Jr, Brown EA, et al: Plasma inorganic fluoride levels with sevoflurane anesthesia in morbidly obese and nonobese patients. Anesth Analg 1993; 76: 1333
187. Thwaites A, Edmends S, Smith I: Inhalation induction with sevoflurane: A double-blind comparison with propofol. Br J Anaesth 1997; 78: 356
188. Muzi M, Colinco MD, Robinson BJ, et al: The effects of premedication on inhaled induction of anesthesia with sevoflurane. Anesth Analg 1997; 85: 1143
189. Muzi M, Robinson BJ, Ebert TJ, et al: Induction of anesthesia and tracheal intubation with sevoflurane in adults. Anesthesiology 1996; 85: 536
190. Tanaka S, Tsuchida H, Nakabayashi K, et al: The effects of sevoflurane, isoflurane, halothane, and enflurane on hemodynamic responses during an inhaled induction of anesthesia via a mask in humans. Anesth Analg 1996; 82: 821
191. Mostafa SM, Atherton AMJ: Sevoflurane for difficult tracheal intubation. Br J Anaesth 1997; 79: 392
192. Eger EI II, Johnson BH: Rates of awakening from anesthesia with I-653, halothane, isoflurane, and sevoflurane: A test of the effect of anesthetic concentration and duration in rats. Anesth Analg 1987; 66: 977
Editors: Barash, Paul G.; Cullen, Bruce F.; Stoelting, Robert K.; Cahalan, Michael K.; Stock, M. Christine