Samir Parekh, MD
![]() What gene is most commonly associated with hereditary hemochromatosis (HHC) and how is it inherited?
What gene is most commonly associated with hereditary hemochromatosis (HHC) and how is it inherited?
Approximately 90% of patients with HHC have mutations in the HFE gene located on the short arm of chromosome 6. The disease has an autosomal recessive inheritance pattern, and the most common point mutation is homozygosity for C282Y. To a much lesser degree, iron overload can occur in compound heterozygotes who have one copy of C282Y and one copy of H63D or S65C.
![]() What is the pathophysiology of HHC?
What is the pathophysiology of HHC?
Mutations in the HFE gene lead to decreased production of hepcidin, a peptide synthesized in the liver that plays a central role in iron regulation. Low concentrations of hepcidin result in increased intestinal absorption of iron without feedback inhibition, and subsequently iron accumulation in the liver and other organs.
![]() What values of transferrin saturation and ferritin should prompt further investigation for hemochromatosis?
What values of transferrin saturation and ferritin should prompt further investigation for hemochromatosis?
Transferrin saturation ≥ 45% and elevated ferritin (typically > 200 µg/L for females and > 300 µg/L for males).
![]() How is the transferrin saturation calculated?
How is the transferrin saturation calculated?
Serum iron/total iron binding capacity × 100%. An increase in transferrin saturation is the earliest laboratory finding in HHC.
![]() When is the serum iron level falsely elevated?
When is the serum iron level falsely elevated?
After meals and at night. A fasting serum iron level collected in the morning is most useful.
![]() True/False: The serum ferritin level is both more sensitive and specific than transferrin saturation values.
True/False: The serum ferritin level is both more sensitive and specific than transferrin saturation values.
False. Ferritin is also an acute phase reactant.
![]() What hepatic conditions may also cause an elevated ferritin level?
What hepatic conditions may also cause an elevated ferritin level?
Chronic viral hepatitis (including Hepatitis C), alcoholic liver disease, and non-alcoholic steatohepatitis.
![]() True/False: In patients with elevated transferrin saturation and ferritin, the next step in the evaluation for HHC is liver biopsy.
True/False: In patients with elevated transferrin saturation and ferritin, the next step in the evaluation for HHC is liver biopsy.
False. The next step in testing for HHC should be HFE gene testing.
![]() True/False: A negative HFE gene test rules out a diagnosis of HHC.
True/False: A negative HFE gene test rules out a diagnosis of HHC.
False. Approximately 10% of HHC patients may be HFE negative. Recently, several other genes and proteins of iron metabolism have been identified as the causes of HHC, although much rarer than HFE-related hemochromatosis. These include mutations in ferroportin (ferroportin disease), transferrin receptor 2 (TfR2 hemochromatosis), and hemojuvelin (HJV hemochromatosis).
![]() In what scenarios should a liver biopsy be considered in HHC?
In what scenarios should a liver biopsy be considered in HHC?
In patients with elevated transaminases or ferritin levels > 1000 µg/L and age > 40, a liver biopsy might be helpful to rule out advanced fibrosis or cirrhosis. In addition, a liver biopsy is useful when the diagnosis is in doubt or non-HFE hemochromatosis is suspected.
![]() What stain is used to examine liver tissue for iron?
What stain is used to examine liver tissue for iron?
Perl’s Prussian blue stain.
![]() True/False: In patients with genetic hemochromatosis, excess iron deposition is found predominantly in parenchymal cells (hepatocytes) with very little iron in cells of the reticuloendothelial (RE) system.
True/False: In patients with genetic hemochromatosis, excess iron deposition is found predominantly in parenchymal cells (hepatocytes) with very little iron in cells of the reticuloendothelial (RE) system.
True. This differs from other, secondary, causes of iron overload.
![]() How do you calculate the hepatic iron index (HII)?
How do you calculate the hepatic iron index (HII)?
The HII is calculated by taking the hepatic iron concentration and dividing by the patient’s age in years. Keep in mind that the hepatic iron concentration must be in µ mol iron/g of dry weight of liver. Remember that the molecular weight of iron is 56.0 because the hepatic iron concentration may be reported in µg iron/g of dry weight of liver. A value > 1.9 is consistent with homozygous HHC. A value < 1.5 is not due to homozygous HHC.
![]() What is the typical HII of a patient with alcoholic liver disease? What is a normal index value?
What is the typical HII of a patient with alcoholic liver disease? What is a normal index value?
1.1 to 1.6; < 0.7 to 1.1.
![]() Patients with ineffective erythropoiesis who require transfusions will have iron deposition in which liver cell populations?
Patients with ineffective erythropoiesis who require transfusions will have iron deposition in which liver cell populations?
In both RE cells and parenchymal cells. Secondary iron overload predominantly affects storage of iron in the RE cells.
![]() Which of the organ(s) involved in genetic hemochromatosis will not improve with phlebotomy?
Which of the organ(s) involved in genetic hemochromatosis will not improve with phlebotomy?
Patients with advanced cirrhosis, arthropathy, and hypogonadism do not improve with therapy.
![]() Which members of a family should be screened for genetic hemochromatosis when it has been diagnosed in one member?
Which members of a family should be screened for genetic hemochromatosis when it has been diagnosed in one member?
The siblings of the proband and all first-degree relatives.
![]() What is the best screening test to identify relatives of individuals with genetic hemochromatosis?
What is the best screening test to identify relatives of individuals with genetic hemochromatosis?
For siblings, HFE gene testing. For children, one option is to perform the HFE gene test on the other parent and screen the children only if the C282Y or H63D gene is present.
![]() Recall all the possible rheumatoid conditions associated with HHC.
Recall all the possible rheumatoid conditions associated with HHC.
Arthropathies involving the second and third metacarpophalangeal joints, joint space narrowing, chondrocalcinosis, subchondral cyst formation, osteopenia, and joint swelling.
![]() What infections are more common in iron-loaded patients?
What infections are more common in iron-loaded patients?
• Vibrio vulnificus
• Listeria monocytogenes, and
• Pasteurella pseudotuberculosis.
![]() How much iron is typically needed to be removed by phlebotomy in a patient with HHC?
How much iron is typically needed to be removed by phlebotomy in a patient with HHC?
10 to 20 g.
![]() How many phlebotomies will this require?
How many phlebotomies will this require?
Each unit of whole blood removed = 250 mg of iron; therefore, at least 40 to 80 units will need to be removed.
![]() How many units of blood should be removed per week to treat patients with HHC?
How many units of blood should be removed per week to treat patients with HHC?
Usually 1 or 2 units/week. After achieving adequate iron depletion, most patients require approximately four maintenance phlebotomies per year.
![]() What are the goal values for transferrin saturation, serum iron, and ferritin during maintenance phlebotomy in patients with HHC?
What are the goal values for transferrin saturation, serum iron, and ferritin during maintenance phlebotomy in patients with HHC?
Transferrin saturation < 50%, low serum iron level, and ferritin < 50 µg/L.
![]() The presence of which two hemochromatosis-related conditions decreases life expectancy compared to the general public?
The presence of which two hemochromatosis-related conditions decreases life expectancy compared to the general public?
Diabetes and/or cirrhosis.
![]() True/False: Due to the low risk of hepatocelluar carcinoma (HCC) in patients with HHC and cirrhosis, HCC screening is not recommended for this patient population.
True/False: Due to the low risk of hepatocelluar carcinoma (HCC) in patients with HHC and cirrhosis, HCC screening is not recommended for this patient population.
False. 30% of patients with HHC and cirrhosis develop HCC representing a 200-fold increased risk. Therefore, these patients should be aggressively screened for HCC.
![]() True/False: The survival rate in patients with genetic hemochromatosis following orthotopic liver transplantation (OLT) is comparable to that of patients who undergo OLT for other indications.
True/False: The survival rate in patients with genetic hemochromatosis following orthotopic liver transplantation (OLT) is comparable to that of patients who undergo OLT for other indications.
True. In the past, patients with HHC had a significantly decreased survival after transplant compared to other patients because of cardiac arrhythmias related to iron overload in the myocardium and infectious complications. Currently, survival rates are comparable due to earlier diagnosis of HHC and iron depletion through phlebotomies prior to transplant.
![]() What is the least likely liver disease to also cause hepatocellular carcinoma?
What is the least likely liver disease to also cause hepatocellular carcinoma?
Wilson’s disease.
![]() True/False: Children less than age 3 should be screened for Wilson’s disease.
True/False: Children less than age 3 should be screened for Wilson’s disease.
False. Clinical manifestations are rarely, if ever, seen before age 5.
![]() In addition to the liver, brain, and joints, what other vital organ can be affected by Wilson’s disease?
In addition to the liver, brain, and joints, what other vital organ can be affected by Wilson’s disease?
Although the heart, pancreas, and eyes have been reported to be involved, the most important other vital organ involved is the kidney. Wilson’s disease can cause Fanconi’s syndrome resulting in low serum phosphorus and uric acid, and failure to excrete acid in the urine.
![]() True/False: A gene for Wilson’s disease has been determined.
True/False: A gene for Wilson’s disease has been determined.
True. ATP7B localized to human chromosome 13. Over 300 different mutations of the ATP7B gene have been identified.
![]() True/False: The Wilson’s disease gene is expressed in the liver and kidney.
True/False: The Wilson’s disease gene is expressed in the liver and kidney.
True. It is also expressed in the brain, lungs, and placenta.
![]() What causes Wilson’s disease?
What causes Wilson’s disease?
There is a gene mutation in the major copper transport protein, ATP7B found in hepatocytes that results in decreased biliary excretion of copper and toxic accumulation in the liver, brain, cornea, and other organs.
![]() What other gastrointestinal disease is associated with copper storage overload and what molecular mechanism is defective?
What other gastrointestinal disease is associated with copper storage overload and what molecular mechanism is defective?
Menke’s disease affects the copper transporter in the proximal small intestine and results in hyperabsorption of copper.
![]() True/False: Wilson’s disease is inherited in a Mendelian fashion as an autosomal dominant allele.
True/False: Wilson’s disease is inherited in a Mendelian fashion as an autosomal dominant allele.
False. Although Wilson’s disease is inherited in a Mendelian fashion, it is inherited as an autosomal recessive allele.
![]() Name two extrahepatic manifestations that often accompany acute liver failure due to Wilson’s disease.
Name two extrahepatic manifestations that often accompany acute liver failure due to Wilson’s disease.
Renal failure and hemolytic anemia.
![]() How is the diagnosis of Wilson’s disease made?
How is the diagnosis of Wilson’s disease made?
The diagnosis requires a constellation of findings including at least two of the following: 1) Low ceruloplasmin level, 2) presence of Kayser–Fleischer rings, 3) typical neurologic symptoms, and 4) liver biopsy with a hepatic copper content of 250 µg per gram of dry weight.
![]() True/False: The serum ceruloplasmin is always decreased (ie, < 20 g/L) in patients with Wilson’s disease.
True/False: The serum ceruloplasmin is always decreased (ie, < 20 g/L) in patients with Wilson’s disease.
False. Remember that up to 5% of Wilson’s patients will have low-normal range of serum ceruloplasmin levels. In addition, a ceruloplasmin level > 30 essentially excludes the disease.
![]() True/False: Only half of patients with Wilson’s disease and liver involvement have the clinical finding shown in the accompanying figure.
True/False: Only half of patients with Wilson’s disease and liver involvement have the clinical finding shown in the accompanying figure.
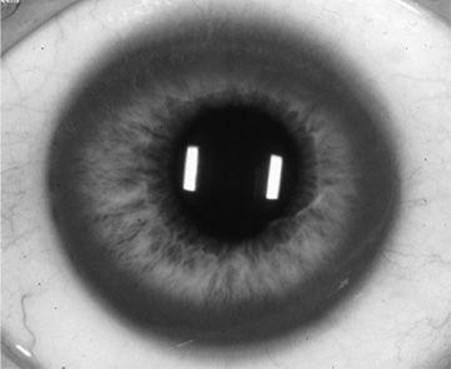
Figure 53-1 See also color plate.
True. This compares to approximately 95% of patients who have neurologic involvement. The figure shows a slit-lamp examination demonstrating the classical Kayser–Fleischer ring which are brownish or gray-green rings that represent fine pigmented granular deposits of copper in Descemet’s membrane in the cornea close to the endothelial surface.
![]() What foods are to be avoided in patients with Wilson’s disease?
What foods are to be avoided in patients with Wilson’s disease?
Organ meats, shellfish, nuts, chocolate, and mushrooms, all of which are high in copper.
![]() Up to 30% of patients with Wilson’s disease develop a side effect of D-penicillamine that necessitates a change of treatment. What are the most common side effects that would necessitate stopping treatment?
Up to 30% of patients with Wilson’s disease develop a side effect of D-penicillamine that necessitates a change of treatment. What are the most common side effects that would necessitate stopping treatment?
Dermatologic-rashes, pemphigus, nephrotic syndrome, Goodpasture syndrome, myasthenia syndrome, aplastic anemia (rare), leukopenia, thrombocytopenia, and systemic lupus erythematosus-like syndrome. Gastrointestinal side effects are most common but usually mild.
![]() What are alternative treatments to D-penicillamine for Wilson’s disease?
What are alternative treatments to D-penicillamine for Wilson’s disease?
Trientine, zinc, and ammonium tetrathiomolybdate.
![]() What other drug must be administered with D-penicillamine?
What other drug must be administered with D-penicillamine?
Pyridoxine (vitamin B6, 25 mg/day).
![]() True/False: D-penicillamine should be stopped during pregnancy.
True/False: D-penicillamine should be stopped during pregnancy.
False. Therapy must be continued but at a lower dose. Stopping therapy can result in significant exacerbations.
![]() True/False: D-penicillamine therapy should be continued for life.
True/False: D-penicillamine therapy should be continued for life.
True. After initial high doses, lower maintenance doses (0.75 to 1 g/day) are instituted.
![]() How is screening performed in the siblings of Wilson’s disease patients?
How is screening performed in the siblings of Wilson’s disease patients?
Serum copper and ceruloplasmin measurements, 24-hour urinary copper measurement and a slit-lamp examination. Children younger than 5 or 6 are usually not affected and should be rechecked at intervals over the next 5 to 10 years. If the mutations of the index case are known, genetic testing should be performed. Siblings have a 25% chance of having the disease.
![]() True/False: Severe neurologic manifestations of Wilson’s disease are an accepted indication for liver transplantation.
True/False: Severe neurologic manifestations of Wilson’s disease are an accepted indication for liver transplantation.
False. Given the uncertain improvement and outcome in neurologic symptoms posttransplant, this is not an indication. Indications for liver transplant include acute liver failure and end-stage liver disease unresponsive to medical therapy. Liver transplantation is curative as it corrects the underlying metabolic defect of the disease.
![]() What is the normal phenotype for the alleles expressing the α1-antitrypsin protease inhibitor (Pi)?
What is the normal phenotype for the alleles expressing the α1-antitrypsin protease inhibitor (Pi)?
PiMM is normal and PiZZ results in the lowest levels of α1-antitrypsin.
![]() How common is α1-antitrypsin deficiency?
How common is α1-antitrypsin deficiency?
α1-antitrypsin deficiency occurs in approximately 1 in 2000 individuals.
![]() Where is the abnormal gene located in α1-antitrypsin?
Where is the abnormal gene located in α1-antitrypsin?
The gene, located on chromosome 14, results in the single amino acid substitution of glutamate by lysine at position 342 leading to a deficiency in sialic acid.
![]() What is the pathophysiology of α1-antitrypsin that leads to liver disease?
What is the pathophysiology of α1-antitrypsin that leads to liver disease?
The gene mutation results in an abnormal folding of the α1-antitrypsin protein and failure of secretion from the hepatocyte endoplasmic reticulum. The defective protein accumulates in the hepatocyte and forms polymers, leading to cellular stress and liver damage.
![]() True/False: Cirrhosis occurs in less than 20% of patients with the PiZZ phenotype.
True/False: Cirrhosis occurs in less than 20% of patients with the PiZZ phenotype.
True. The PiZZ phenotype, in several studies, caused cirrhosis in only 12% of patients. In contrast, chronic obstructive pulmonary disease occurs in roughly 75% of these patients.
![]() Do other α1-antitrypsin phenotypes, that is, heterozygotes, result in chronic liver disease?
Do other α1-antitrypsin phenotypes, that is, heterozygotes, result in chronic liver disease?
It must be remembered that certain heterozygous states can result in chronic liver disease. For instance, patients with PiSZ and PiZZ can develop cirrhosis. MZ heterozygotes usually do not develop disease unless there is some other superimposed liver condition, such as alcoholic liver disease or chronic viral hepatitis. Liver disease due to other causes may progress more rapidly in individuals who have an MZ phenotype.
![]() What is an effective treatment for patients with α1-antitrypsin deficiency?
What is an effective treatment for patients with α1-antitrypsin deficiency?
The only treatment for α1-antitrypsin-related liver disease is symptomatic management of complications and liver transplantation. With liver transplantation, the phenotype becomes that of the transplanted liver. Recall that the liver is the site of production of this protease inhibitor, and therefore, liver transplantation is curative. α1-antitrypsin replacement therapy has no effect on liver disease since liver injury is related to accumulation of the mutant protein within the hepatocyte and not a lack of circulating antiproteases.
![]() True/False: The α1-antitrypsin level is the best test to detect deficiency states associated with cirrhosis.
True/False: The α1-antitrypsin level is the best test to detect deficiency states associated with cirrhosis.
False. The best diagnostic test is obtaining the phenotype. The level may be low-normal even in states of homozygous deficiency. Also, α1-antitrypsin is an acute phase reactant and levels may rise in the setting of illness or other types of inflammatory stress.
![]() What is the characteristic finding of α1-antitrypsin deficiency on liver biopsy?
What is the characteristic finding of α1-antitrypsin deficiency on liver biopsy?
Presence of eosinophilic, periodic acid-Schiff-positive, and diastase-resistant globules in the endoplasmic reticulum of periportal hepatocytes.
![]() What is the frequency of genetic hemochromatosis, of Wilson’s disease, and of α1-antitrypsin deficiency?
What is the frequency of genetic hemochromatosis, of Wilson’s disease, and of α1-antitrypsin deficiency?
Genetic hemochromatosis (1 in 250 individuals); Wilson’s disease (1 in 30,000 individuals); α1-antitrypsin deficiency (1 in 2000 individuals).
![]() What is the most common of the acute porphyrias?
What is the most common of the acute porphyrias?
Acute intermittent porphyria (AIP) occurs in 5 to 10 per 100,000 people. Its inheritance pattern is autosomal dominant with incomplete penetrance.
![]() What is the enzyme deficiency in AIP?
What is the enzyme deficiency in AIP?
There is a 50% reduction in porphobilinogen (PBG) deaminase activity.
![]() What are the major manifestations of AIP?
What are the major manifestations of AIP?
Derangements in the autonomic nervous system.
![]() What are the predominant heme by-products in the urine of a patient with an acute attack of AIP?
What are the predominant heme by-products in the urine of a patient with an acute attack of AIP?
PBG and 5-aminolevulinic acid (ALA). PBG quantities are higher than ALA. These levels may be normal between attacks.
![]() True/False: AIP is the only porphyria that is not associated with cutaneous manifestations.
True/False: AIP is the only porphyria that is not associated with cutaneous manifestations.
False. AIP is one of two acute porphyrias with only neurologic findings. The other is ALA dehydratase deficiency.
![]() What precipitates episodes of the acute porphyrias?
What precipitates episodes of the acute porphyrias?
Prescription or recreational drugs, particularly corticosteroids and derivative hormones. This is why diagnosis is, oftentimes, first made at puberty. Alcohol ingestion, smoking, fasting, infection, stress, and pregnancy are other risk factors.
![]() True/False: All of the heme synthetic enzymes are expressed only in the liver.
True/False: All of the heme synthetic enzymes are expressed only in the liver.
False. Three enzyme deficiencies among the cutaneous porphyrias are expressed in the bone marrow.
![]() What is the most common of the porphyrias?
What is the most common of the porphyrias?
Porphyria cutanea tarda (PCT) is the most common of the porphyrias, usually presenting after 10 years of age.
![]() What do AIP and PCT have in common?
What do AIP and PCT have in common?
Enzyme expression occurs only in the liver, both have autosomal dominant patterns of inheritance (the former with incomplete penetrance while the latter can be acquired), and both are the most common (the former being acute while the latter being of the cutaneous porphyrias).
![]() What is the typical lesion associated with PCT?
What is the typical lesion associated with PCT?
Photosensitivity-induced vesicles and bullous lesions, or blisters.
![]() PCT is strongly associated with what other disorders?
PCT is strongly associated with what other disorders?
Excess alcohol intake, estrogen therapy, systemic lupus erythematosus, diabetes mellitus, chronic renal failure, acquired immunodeficiency syndrome, and chronic hepatitis C. Of note, most patients also have iron overload.
![]() True/False: Patients with acute porphyrias are at increased risk of developing hepatocellular carcinoma.
True/False: Patients with acute porphyrias are at increased risk of developing hepatocellular carcinoma.
True. Even though hepatic involvement is variable and mild.
![]() What two porphyrias are most commonly associated with liver complications?
What two porphyrias are most commonly associated with liver complications?
PCT and hepatoerythropoietic porphyria (HEP).
![]() What clinical clues should lead to consideration of the diagnosis of porphyria?
What clinical clues should lead to consideration of the diagnosis of porphyria?
Recurrent bouts of severe abdominal pain, constipation, neuropsychiatric disturbances, and typical dermatologic findings.
![]() How do you treat PCT?
How do you treat PCT?
Avoid precipitating factors (alcohol, sun, etc). Phlebotomy is considered the standard of care. Hydroxychloroquine may also be useful. If possible, HCV treatment should be considered but usually after PCT is adequately treated.
![]() What are the two bile acid transport disorders in which a genetic defect of primary bile acid secretion is believed responsible?
What are the two bile acid transport disorders in which a genetic defect of primary bile acid secretion is believed responsible?
Byler’s syndrome and Alagille’s syndrome.
![]() What is the hepatic lesion associated with cystic fibrosis?
What is the hepatic lesion associated with cystic fibrosis?
Focal biliary cirrhosis. Over time, focal biliary cirrhosis can progress to multilobular biliary cirrhosis and clinically significant portal hypertension.
![]() True/False: No medical therapy has been shown to be of benefit in patients with cystic fibrosis-related liver disease.
True/False: No medical therapy has been shown to be of benefit in patients with cystic fibrosis-related liver disease.
False. Controlled studies have demonstrated the beneficial effects of ursodeoxycholic acid in terms of improvement in cholestasis and nutritional status.
• • • SUGGESTED READINGS • • •
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and Management of Hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;5(1):328-343.
Roberts EA, Schilsky ML. Diagnosis and Treatment of Wilson Disease: An Update. Hepatology. 2008;47(6):2098-2111.