Sheila N. J. Sait
Maria R. Baer
Cytogenetics is the study of chromosome structure. Cytogenetic analysis has become increasingly important in the diagnosis, classification, management, and scientific investigation of hematopoietic and lymphoid disorders. Classical cytogenetic analysis studies cells in metaphase, or dividing cells. However, with the advent of molecular cytogenetic techniques such as fluorescence in situ hybridization, cytogenetic analysis now also includes the study of interphase, or nondividing, cells.
Cytogenetic techniques established over the last five decades have become a routine part of the analysis of abnormal hematopoiesis and lymphopoiesis, and hence play a prominent role in the clinical practice of hematology and oncology. The utility of cytogenetics in the diagnosis and management of each disease depends on selection of the appropriate tissue and cell type for analysis, availability of cells for karyotyping, and accurate interpretation of cytogenetic findings.
From a scientific point of view, the identification of recurring chromosomal abnormalities has guided the search for genes involved in malignant transformation and has helped to elucidate alterations in the structure and expression of these genes. Identification of genes involved and elucidation of their altered structure and expression have in turn led to an understanding of mechanisms of malignant transformation and are now fostering the development of targeted therapies.
This chapter focuses on general principles of cytogenetics, cytogenetic methods, terminology, and the application of cytogenetic analysis in hematology and hematologic oncology. Cytogenetic findings in specific diseases are also discussed in detail in the chapters devoted to those diseases.
![]() Background
Background
Because cytogenetic analysis is the study of chromosome structure, it requires that chromosomes be visualized microscopically as discrete structures. Every human cell contains DNA packaged into chromosomes, normally consisting of 22 pairs of autosomes and one pair of sex chromosomes. Chromosome morphology varies markedly during the cell cycle, which is the process of cell replication.
The cell cycle consists of interphase, the period between cell divisions, and mitosis, or cell division. Cells that are in a resting state and not cycling are said to be in G0.
Interphase is the portion of the cell cycle in which the nucleus is most metabolically active and undergoes chromosome duplication in preparation for mitosis. Interphase includes three distinct stages: The G1 (gap one) phase, the S (synthesis of DNA) phase, and the G2 (gap two) phase.
Mitosis (M phase), or cell division, is divided into five phases: Prophase, prometaphase, metaphase, anaphase, and telophase. During prophase the chromatin, which has already doubled, undergoes progressive coiling and condensation into well-defined chromosomes. In prometaphase, the nuclear membrane breaks down, releasing the chromosomes into the cytoplasm, and the chromosomes move to the center of the cell or the equatorial plane. During metaphase the chromosomes line up in the center or metaphase plate. This is when the chromosomes are most contracted and least metabolically active. Anaphase is when the centromere divides, leading to the separation of the chromatids (sister chromosomes). Once separated, the chromatids move to the opposite ends of the cell. Telophase, the final stage of mitosis, is when the chromosomes uncoil, the nuclear membrane reappears around each set of chromosomes, and the nucleus takes on the morphology seen in interphase. During, or directly after, telophase, new cell membranes (cytokinesis) are formed and divide the cytoplasm, creating two identical daughter cells.
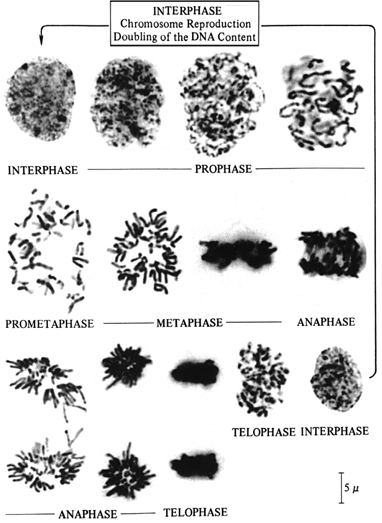
The appearance of cells during the different phases of the cell cycle is shown in Figure 3.1. Interphase chromosomes are uncoiled and cannot be resolved as discrete structures. Chromosomes first become visible as discrete structures during prophase. Prometaphase chromosomes are almost maximally condensed and are visualized as two identical chromatids. Maximal chromosome condensation occurs during metaphase. The chromosomes then become less condensed during anaphase and telophase; individual chromosomes can no longer be visualized. Cytogenetic analysis must be performed on cells in prometaphase or metaphase.
Successful analysis of chromosome number and morphology depends on many factors. Adequate numbers of cells must be available. Cells must be viable and undergoing division. Sufficient numbers of cells must be present in the phases of the cell cycle with chromosome morphology appropriate for analysis. Chromosomes must be separated from one another within the cell so that each chromosome can be resolved as a distinct entity. Finally, each chromosome must be able to be identified and characterized as normal or abnormal. For cytogenetic analysis of malignancies, it is critical that malignant cells be analyzed, rather than coexisting normal cells.
![]() History
History
The ability to study chromosomes depended on the development of methods for growing cells in tissue culture, arresting cells at the appropriate phases of the cell cycle, separating chromosomes from each other within cells, and optimizing their morphology and staining.
The presence of 46 chromosomes in normal human cells was first demonstrated in 1956 (1,2). Normal male cells were found to contain two copies of each of 22 autosomes and one copy of each of the sex chromosomes, X and Y, while normal female cells contain two copies of each of the 22 autosomes and of the X chromosome, and no Y chromosome.
Associations between abnormal numbers of chromosomes and congenital anomalies were initially reported in 1959. Three, rather than two, copies of one of the smallest human chromosomes were found in cells of children with Down syndrome (3). Presence of two copies of the X chromosome, along with one copy of the Y chromosome, was reported in cells of males with Klinefelter syndrome (4), and a single copy of the X chromosome was found in cells of females with Turner syndrome, without a Y chromosome (5).
In 1960, Nowell and Hungerford reported the first chromosomal anomaly associated with cancer (6). The presence of an unusually small chromosome was detected as a consistent abnormality in bone marrow metaphase spreads from patients with chronic myeloid leukemia (CML) (Fig. 3.2). This small chromosome was called the Philadelphia chromosome, after the city in which it was discovered. The Philadelphia chromosome was initially abbreviated Ph1; the designation Ph1 was subsequently changed to Ph (7).
|
|
|
Figure 3.1. Mitotic cycle in cultured human lymphocytes. (From Therman E, Susman M. Human chromosomes. 3rd ed. New York: Springer-Verlag.) |
|
|
|
Figure 3.2. An unbanded metaphase spread from bone marrow cells of a patient with chronic myelogenous leukemia, with the Philadelphia chromosome (arrow). (From Sandberg AA. The chromosomes in human cancer and leukemia. 2nd ed. New York: Elsevier, 1956.) |
In the late 1960s, staining of metaphase spreads with quinacrine mustard was found to produce unique patterns of fluorescence on each chromosome (8). A human karyotype banded with this first chromosome banding technique was first published in 1970 (9). However quinacrine banding (Q-banding) required fluorescence microscopy and could be cumbersome, as the fluorescence tended to “quench” rapidly. Other banding techniques subsequently developed produced similar bands using pretreatment with alkali and saline followed by staining with Giemsa, a compound that had been developed to stain blood smears for the identification of the protozoan that causes malaria (10). Giemsa banding (G-banding) could be visualized by light microscopy and became the most commonly used banding technique. Chromosome band nomenclature was established in 1971 (7).
Banding allows chromosomes to be visualized as a continuous series of light and dark regions. Bands are defined as regions of chromosomes that are distinguished from adjacent regions by virtue of appearing darker or lighter with one or more banding techniques. Unique banding patterns allow chromosomes to be distinguished from one another. Identification of individual chromosomes had not previously been possible.
Chromosome banding allowed more precise identification and description of cytogenetic abnormalities. In 1973, Rowley reported that the recurring cytogenetic abnormality in CML was a reciprocal translocation between chromosomes 9 and 22, t(9;22)(q34;q11) (11). The Philadelphia chromosome was an abnormally small chromosome 22 resulting from the translocation with chromosome 9. The use of chromosome banding also rapidly increased the numbers of recurring cytogenetic abnormalities reported. The t(8;21) in acute myeloid leukemia (AML) was reported in 1973 (12) and the t(15;17) in acute promyelocytic leukemia (APL) in 1977 (13).
The late 1970s saw the development of high-resolution banding (14), achieved by analyzing prophase and prometaphase chromosomes, which are more elongated, thus allowing visualization of 500 to 1,000 bands, whereas metaphase banding yields only approximately 300 bands. Availability of sufficiently large numbers of prophase and prometaphase cells for analysis is achieved by cell cycle synchronization. Cells are cultured with methotrexate, which arrests DNA synthesis by inhibiting dihydrofolate reductase, the enzyme that catalyzes conversion of folic acid to tetrahydrofolic acid, a coenzyme for one-carbon unit transfers including methylation of deoxyuridylic acid to thymidylic acid. Inhibition of DNA synthesis arrests the cell cycle in interphase. The cell cycle block is subsequently released by transferring the cells to methotrexate-free, thymidine-rich medium. Cells enter mitosis simultaneously and are arrested in prophase and prometaphase by brief colchicine treatment. High-resolution banding allows identification of abnormal karyotypes in larger percentages of cases (15), and recognition of subtle structural chromosomal abnormalities such as inv(16)(p13q22), a recurring abnormality in AML (16).
The last 20 years have seen a very rapid expansion of the number of recurring cytogenetic abnormalities reported in malignancies. Cytogenetic databases are now available online. The two most commonly used are the Mitelman Database of Chromosome Aberrations in Cancer (Mitelman F, Johansson B, Mertens F, eds., http://cgap.nci.nih.gov/Chromosomes/Mitelman) and the Atlas of Genetics and Cytogenetics in Oncology and Haematology (http://www.infobiogen.fr/services/chromcancer/). Large numbers of cytogenetic abnormalities have been described in hematologic malignancies, while data on cytogenetic abnormalities in lymphomas and solid tumors are more limited, reflecting the greater difficulty of obtaining analyzable dividing cells from lymph nodes and tumor tissue, compared to bone marrow samples.
In the 1980s molecular technology was combined with the conventional cytogenetic methodology described above to create molecular cytogenetic techniques, or in situ hybridization (ISH). The most widely used technique is fluorescence in situ hybridization (FISH), in which specific DNA or RNA sequences indirectly or directly labeled with fluorochromes are hybridized to complementary target sequences in metaphase or interphase cells, and are subsequently visualized by fluorescence microscopy (17,18). Locus-specific probes are used to detect the presence and location of a particular gene or unique DNA sequence. Centromeric probes are highly repetitive sequences that detect α satellite DNA primarily located in the centromeres. Subtelomeric probes contain unique sequences from the telomere region of chromosomes. FISH can be performed simultaneously with different probes labeled with different fluorochromes, permitting the simultaneous detection of several target sequences. Finally, whole chromosome paints are pooled DNA probes to numerous unique and repetitive sequences homologous to many sites on sorted single chromosomes, allowing “painting” of each chromosome (19). Since these chromosome-specific probes label entire chromosomes, they are particularly useful in identifying the origin of chromosomal material that is not identifiable in banded metaphases. Applications of FISH are illustrated in the sections of this chapter that focus on numerical and structural chromosomal abnormalities.
Specialized imaging procedures have allowed simultaneous visualization of all the chromosomes in a cell. These techniques, called multiplex fluorescence in situ hybridization (M-FISH) (20) and spectral karyotyping (SKY) (21), utilize combinatorial labeling and digital imaging microscopy to create a unique color for each chromosome, so that all chromosomes, each with unique labeling, can be visualized simultaneously. These techniques are extremely useful in identifying the chromosomal origins of marker chromosomes and otherwise unidentified genetic material.
Competitive genomic hybridization (CGH), described in the early 1990s, is a molecular cytogenetic technique that allows efficient screening of cells for losses and gains of chromosomal material (22,23). Genomic DNA from the cells to be studied (test DNA) is labeled with a fluorochrome, and DNA from normal cells (control DNA) is labeled with a different color fluorochrome. Labeled test DNA and genomic DNA are simultaneously hybridized to metaphase chromosome spreads from normal cells. The fluorescent signals generated by the test DNA and the control DNA along the normal chromosomes are compared as a means of detecting and identifying chromosomal material that has been lost or gained in the test DNA. Computerized image analysis is used to measure and compare signal intensities.
The most recent innovation has been the introduction and increasing use of microarray-based CGH or array CGH, which allows the detection of small genomic imbalances including deletions, duplications, and copy number variations that cannot be detected by conventional chromosome analysis and traditional CGH. Array CGH uses differentially labeled test and reference genomic DNA that are simultaneously hybridized to DNA targets arrayed on a solid platform, thus improving the resolution of the assay.
![]() Cytogenetic Methods
Cytogenetic Methods
Samples
Conventional cytogenetic analysis must be performed on tissues that are sources of dividing cells. For successful cytogenetic analysis of malignancies, it is critical that the cells studied be the malignant cells, rather than contaminating normal cells. Bone marrow is the tissue of choice for cytogenetic analysis of leukemias, myelodysplastic syndromes (MDSs), and myeloproliferative disorders (MPDs). In cases of leukemia in which marrow cannot be aspirated, blood may be used if it contains >10% blasts. A bone marrow core biopsy sample can sometimes be processed successfully to allow study of cells in mitosis. Blood is generally not appropriate for cytogenetic analysis of MDS.
A sample of 1 to 2 ml of bone marrow is generally adequate for cytogenetic analysis. The sample must be aspirated into a sterile syringe coated with preservative-free sodium heparin to prevent clotting and then transferred into a sterile tube containing preservative-free heparin or a medium such as RPMI 1640, McCoy’s 5A, or Hanks’ balanced salt solution with preservative-free heparin. Antibiotics (penicillin, streptomycin) may be added to the transport medium. When peripheral blood is to be studied, 10 ml of blood should be drawn aseptically into a sterile heparinized syringe and transferred to a sterile tube also containing heparin.
Cytogenetic analysis of lymphomas is performed on lymph node tissue. Lymph nodes obtained aseptically are transported in sterile tubes containing medium and antibiotics and transferred to sterile Petri dishes with medium, where they are minced using sterile scissors and then triturated using a Pasteur pipette to create a cell suspension. Processing of this cell suspension is similar to that of bone marrow and blood cells.
Samples should be processed for cytogenetic analysis as soon as possible after they are obtained. Delays in processing may compromise cell viability. Additionally, normal cells may survive better than malignant cells, so that only normal karyotypes are obtained. Samples with very high numbers of cells are particularly adversely affected by delays in processing (24), as are acute lymphoblastic leukemia (ALL) samples, regardless of cell numbers. If bone marrow or blood samples cannot be processed immediately or must be mailed overnight to be processed, cells should be maintained in culture medium at room temperature. Lymph nodes may be refrigerated. Samples for cytogenetic analysis should never be frozen.
Sample Processing and Cell Culture
Marrow, blood, or lymph node cells are centrifuged and then resuspended in a medium such as RPMI 1640 or McCoy’s 5A. They are then counted with a Coulter counter or a hemocytometer and resuspended at 1 × 106 cells/ml, which is the optimal concentration for cell culture for cytogenetic analysis.
Cytogenetic analysis may be performed on uncultured or cultured cells. Chromosome preparations from cells that have not been cultured are called direct preparations, and preparations from cultured cells are called indirect. Bone marrow cells can be studied in direct preparations because dividing cells are present in bone marrow aspirates. The advantage of direct preparations is that they allow rapid karyotyping of cells that are undergoing division in vivo, but the disadvantages are a low mitotic index because in vivo cell proliferation rates are generally low, and generally poor chromosome morphology. Additionally, compared to indirect preparations, direct preparations more often yield normal rather than abnormal metaphases (25). The normal metaphases seen in direct preparations may derive from erythroblasts (26,27). In APL, in particular, analysis of indirect preparations demonstrates the t(15;17) translocation in virtually all cases (28), but direct preparations often yield normal karyotypes (29). Cell culture increases the mitotic rate, improves chromosome morphology, and generally promotes proliferation of malignant cells, although normal cells may also proliferate in culture. To maximize the likelihood of successfully karyotyping the abnormal population, both direct and indirect preparations should be studied; several culture conditions should be used, including cell cycle synchronization; and cell cultures should be harvested at several different times.
Mitogens may be useful in stimulating proliferation of abnormal cells in some circumstances. In particular, unstimulated chronic lymphocytic leukemia (CLL) cells have a very low mitotic rate, and cytogenetic analysis in cases of CLL studied without mitogens often reveals normal metaphases that are derived from coexisting normal cells, rather than from CLL cells. The use of B-cell mitogens increases the frequency with which cytogenetic abnormalities are detected in cases of CLL. To optimize the success of cytogenetic analysis, B-cell CLL cells should be cultured with a polyclonal B-cell activator such as 12-0-tetradecanoylphorbol-13-acetate (TPA), Epstein–Barr virus (EBV), lipopolysaccharide (LPS), and protein A to stimulate B-cell proliferation (30).
T-cell CLL cells may be cultured with a combination of phytohemagglutinin (PHA), TPA, and pokeweed mitogen to stimulate cell division. CLL cells should be cultured for long periods of time, in the range of 3 to 5 days. The likelihood of successful cytogenetic analysis in cases of multiple myeloma is also increased by cell culture with combinations of cytokines, including granulocyte– macrophage colony-stimulating factor, interleukin-3, and interleukin-6 (31).
Cells are incubated (direct preparations) or cultured (indirect preparations) in a medium such as RPMI 1640 or McCoy’s 5A supplemented with fetal calf serum, L-glutamine, penicillin, and streptomycin; mitogens are added in selected cases. The cell density should be 1 × 106 cells/ml of medium. Colchicine (Colcemid) is used to arrest cells in metaphase. In direct preparations, colchicine is added to the cell suspension without prior cell culture, and cells are harvested after incubation in medium with colchicine at 37°C for 1 hour. In overnight colchicine-treated cultures, cells are cultured overnight in medium with colchicine and are harvested the next morning. In short-term cultures, cells are cultured in medium without colchicine at 37°C, generally for 24 to 72 hours. Before harvesting cells after short-term culture, colchicine is added to the cultures, usually at 0.01 μg or 0.02 μg/ml for 30 minutes to 1 hour. Longer colchicine exposure times result in greater numbers of metaphases, but longer colchicine exposure times and higher colchicine concentrations compromise chromosome morphology.
Cell cycle synchronization in the early stages of cell division yields elongated chromosomes, allowing high-resolution chromosome banding. To achieve cell cycle synchronization, cell division is blocked by incubation with an antimetabolite, and the block is then released, producing synchronization of cell division in early mitosis, when chromosomes are longer than in metaphase. Moreover, because cell division is synchronized, the duration of colchicine treatment can be significantly reduced, resulting in better chromosome morphology. The most commonly used technique is methotrexate (MTX) synchronization. MTX, an inhibitor of thymidine biosynthesis, arrests cells in S phase. Cells are initially cultured in medium without MTX, which is added after the initial culture period. After about 17 hours, most of the dividing cells have accumulated in S phase. At this point, thymidine is added to the culture, releasing the MTX block. The cells then proceed synchronously to mitosis. Colchicine is then added for 10 to 15 minutes, and the cells are harvested. Shortened colchicine treatment, in addition to MTX synchronization, produces elongated chromosomes, allowing detection of subtle abnormalities such as small deletions and rearrangements.
To increase the likelihood of identification and successful cytogenetic analysis of abnormal clones, a variety of culture conditions should be used, including synchronization of cell division. Optimizing cell density and using different culture conditions are particularly important in maximizing the likelihood of successful cytogenetic analysis of ALL cells.
The constitutional karyotype is most commonly established by cytogenetic analysis of stimulated peripheral blood. Peripheral blood cells are cultured at 1 × 106 cells/ml in culture medium supplemented with the T-cell mitogen PHA (2%). Colchicine is added after 72 to 96 hours, generally at a concentration of 0.01 μg/ml. Cultures are harvested after 1 hour of colchicine exposure.
Hypotonic Treatment
Following colchicine treatment, hypotonic treatment is used to produce cell swelling, allowing chromosomes to spread within cells. Chromosome spreading serves to optimize visualization of each chromosome and to minimize overlapping of chromosomes in metaphase spreads. A dilute solution of KCl (0.075 M) is most commonly used for hypotonic treatment; however, other hypotonic solutions are also in use. After colchicine treatment, cells are centrifuged, resuspended in prewarmed hypotonic solution, and incubated at 37°C. The optimal duration of incubation in hypotonic medium varies; it is generally in the range of 10 to 45 minutes.
Fixation
After hypotonic treatment, cells are fixed in a modified Carnoy fixative (3:1 methanol:acetic acid volume/volume). Fixation causes denaturation and precipitation of proteins and nucleic acids, resulting in hardening of chromatin, which improves morphology. The cells are centrifuged, gently resuspended in fixative, incubated at room temperature, then centrifuged again. This is repeated until a cell suspension that is only slightly cloudy is obtained. Slides may be prepared immediately for cytogenetic analysis, or the cells may be stored in fixative and prepared for analysis at a later time.
Slide Preparation
Fixed cells are dropped onto cleaned glass slides under several different conditions. The angle at which the pipette and slide are held is varied. Slides may be wet, cold, or dry, and are then dried with steam, air, or flame. After the slides are dried, they are examined for cell density and quality of metaphase spreads by phase contrast microscopy. If the slides are not optimal, new slides are prepared, altering the density of the cell suspension or the dropping technique, as appropriate.
Staining
Since 1968, several banding techniques have been developed that yield staining of chromosome regions with variable intensity based on their nucleotide and protein composition. Staining with dyes produces a unique banding pattern on each chromosome that is specific for that chromosome.
Q-banding, the first banding method developed, uses quinacrine mustard or quinacrine dihydrochloride to create fluorescent transverse bands when excited with the proper wavelength of light. Quinacrine is a fluorescent dye that intercalates into the DNA helix. Q-band analysis requires fluorescence microscopy, and Q-bands fade over time. Q-banding is not used for routine cytogenetic analysis.
G-banding uses pretreatment with an enzyme (trypsin) followed by staining with Giemsa dye to also produce transverse bands, called G-bands, which are identical to Q-bands. G-banding is generally preferred over Q-banding because G-band analysis is performed by light microscopy and is permanent. G-banding is the most widely used banding technique for routine chromosome analysis. Other G-banding techniques utilize stains other than Giemsa, such as Wright and Leishman stains.
The most widely used G-banding technique is called GTG banding (G-bands by trypsin using Giemsa). In this technique, air-dried slides aged by heating in an oven are treated with a dilute solution of the proteolytic enzyme trypsin (0.05% trypsin for 5 to 10 minutes), and then stained with Giemsa. The relative importance of DNA and proteins in G-banding is not well understood. Treating chromosomes with a hot alkaline solution before Giemsa staining produces bands that are the reverse of G-bands, called R-bands. R-banding methods are useful for analyzing rearrangements involving the terminal ends of chromosomes.
In contrast to Q-, G-, and R-banding, which produce bands distributed along the entire length of each chromosome, other staining methods are adapted to demonstrating specific chromosomal structures, including constitutive heterochromatin (C-banding), telomeric regions (T-banding), and nucleolus-organizing regions (NOR-banding). C-bands are primarily in the centromeric regions of chromosomes. C-band size and position are unique for each chromosome. C-banding is particularly useful in studying chromosome translocations involving centromeric regions.
|
|
|
Figure 3.3. Banded metaphase. (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) |
Microscopy
Following banding, slides are scanned under a light microscope to locate chromosome spreads that are of good quality for karyotyping (Fig. 3.3). The coordinates of these metaphases are recorded.
Adequate cytogenetic analysis requires analysis of at least 20 chromosome spreads. In particular, whereas abnormal clones are sometimes demonstrated by analysis of small numbers of cells, demonstration of 20 normal chromosome spreads is required for a case to be considered cytogenetically normal. Demonstration of 20 out of 20 normal metaphases excludes the presence of 14% abnormal cells with 95% confidence (32).
Cytogenetic analysis of cases of ALL presents particular difficulties. Chromosome spreading is more difficult to accomplish, chromosome morphology is fuzzy, and bands are often suboptimal. Thus, many metaphase spreads cannot be analyzed. Two morphologically different populations of metaphases may be present. When two populations are present, the population with better spreading and better chromosome morphology often consists of cells with normal karyotypes. If the metaphases that are easiest to study are selected for analysis, the case will be inaccurately characterized as cytogenetically normal. Therefore, it is imperative to examine both populations.
Karyotyping
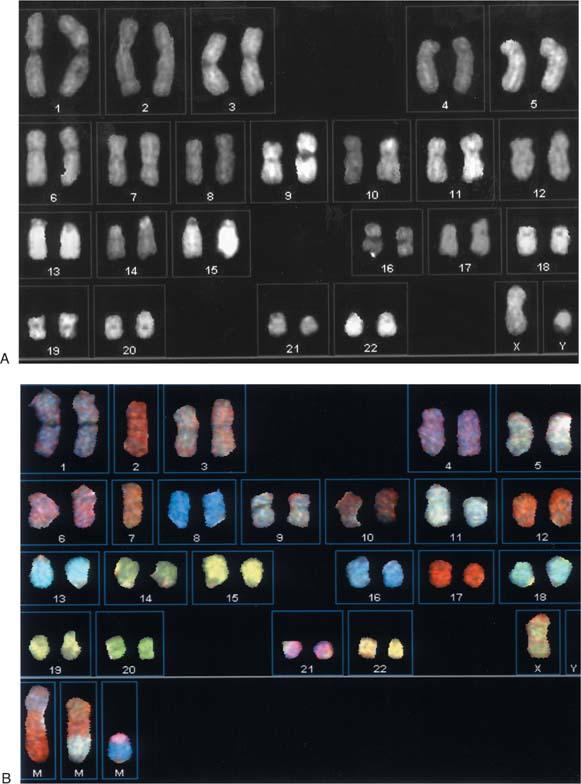
Chromosomes are counted in each chosen metaphase spread to determine whether they are present in normal numbers. Chromosomes are then identified and characterized as structurally normal or abnormal. To facilitate analysis, chromosomes from each metaphase spread are arranged in a prescribed order, called a karyotype (Fig. 3.4A and B).
In recent years, automated karyotyping systems have greatly increased the efficiency of karyotyping in laboratories that handle large numbers of samples. Automated karyotyping relies on computerized image analysis. Software programs have been created that allow chromosome spreads identified by light microscopy to be karyotyped on a computer screen. Karyotypes are then printed directly from the computer. Chromosome spread and karyotype images are archived as digital files. Automated karyotyping obviates the need to photograph metaphases, develop film, print negatives, and cut and paste chromosomes. The number of steps and the amount of time required for cytogenetic analysis are significantly decreased.
The ability to detect subtle cytogenetic abnormalities depends heavily on the experience of the cytogeneticist. Examples of subtle structural abnormalities that may be easily missed by cytogeneticists without extensive experience include inv(16)(p13q22) and t(9;11)(p21;q23), which are clinically significant cytogenetic abnormalities in AML. Accurate karyotyping of metaphases with multiple or complex abnormalities is also a function of experience. Cytogenetic analysis of cancer cells is optimally performed in laboratories staffed by cytogeneticists with expertise in this specialized area.
|
|
|
Figure 3.4. Normal G-banded male (A) and female (B) karyotypes. (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) |
Cytogenetic Nomenclature
Human chromosome nomenclature was initially established at an international conference in 1960. It has been serially updated to include terminology developed to describe chromosomal abnormalities, chromosome bands, high-resolution bands, and, most recently, results of FISH analysis. The most recent guidelines for cytogenetic nomenclature were published in 2005 (7).
For karyotypic analysis, each chromosome is visualized as two chromatids that are joined at a central constriction called the centromere. The centromere is the region where the chromosome attaches to the spindle during mitosis. The centromere divides the chromosome into two arms: A short arm, designated the p-arm (p stands for petit), and a long arm, designated the q-arm (q was chosen simply because it is the letter after p). By convention, chromosomes are always shown with the p-arm on top.
Chromosomes were initially described based on their size and the position of the centromere. Size was characterized as large, medium, or small. The position of the centromere was used to classify chromosomes as metacentric, submetacentric, or acrocentric. Chromosomes with the centromere in the middle are designated as metacentric, those with the centromere closer to one end are submetacentric, and those with the centromere almost at one end are called acrocentric.
In 1963, before the advent of chromosome banding, human chromosomes were divided into seven groups, A through G, based on their size and the position of the centromere. Group A included chromosomes 1, 2, and 3, which are large, metacentric chromosomes. Group B included chromosomes 4 and 5, which are large and submetacentric. Group C chromosomes, including numbers 6 through 12 and the X chromosome, are medium-sized and metacentric or submetacentric. Group D, chromosomes 13 through 15, includes medium-sized acrocentric chromosomes with satellites. Group E, chromosomes 16 through 18, consists of small metacentric (chromosome 16) and submetacentric (chromosomes 17 and 18) chromosomes. Group F chromosomes, numbers 19 and 20, are small and metacentric. Group G chromosomes, including 21, 22, and Y, are small and acrocentric; 21 and 22 have satellites, whereas the Y chromosome does not. Without chromosome banding, chromosomes within some groups could not be distinguished. Banding allows the chromosomes in each group to be uniquely identified and distinguished from each other.
Nomenclature was then established to describe the structure of individual chromosomes. Landmarks are distinct morphologic features important in identifying chromosomes, and include the centromere, the ends of the chromosomes (called telomeres), and prominent chromosome bands. Chromosome regions are defined as areas lying between adjacent landmarks, and range between one and four in number on the short and long arms of individual chromosomes. Regions of each chromosome arm are numbered sequentially, moving outward from the centromere toward the telomere (Fig. 3.5).
Bands are defined as chromosome segments that are clearly distinguishable from adjacent segments by virtue of appearing lighter or darker with one or more banding techniques. Chromosome regions, defined above, are divided into bands. The bands within regions are also numbered sequentially, also moving outward on each arm from the centromere toward the telomere (Fig. 3.5). To designate chromosome bands, one specifies the chromosome number, the chromosome arm, the region number, and the band number within the region, given in order, without spacing or punctuation. For example, 1p31 (chromosome 1, short arm, region 3, band 1) designates the first band of the third region of the short arm of chromosome 1.
High-resolution banding divides chromosome bands into sub-bands, which are light and dark regions that constitute finer structure within chromosome bands. Sub-bands are also numbered from the centromere toward the telomere of each arm. Sub-band numbers are given after band numbers, separated by decimal points. For example, band 1p31 includes sub-bands 1p31.1, 1p31.2, and 1p31.3. Figure 3.5 shows normal human chromosomes with sub-bands as well as bands.
Karyotypes are described according to the International System for Human Cytogenetic Nomenclature (7). The description of a karyotype starts with the total number of chromosomes, including the sex chromosomes. The total number of chromosomes is followed by a list of the sex chromosomes present in the cell, and then a list of abnormalities of autosomes listed in ascending numerical order. This system represents a change from older nomenclature, in which numerical chromosomal abnormalities were listed first, followed by structural abnormalities. A normal male karyotype is 46,XY and a normal female karyotype is 46,XX. Common abbreviations and symbols used in describing karyotypes are listed and defined in Table 3.1.
Chromosome abnormalities may be numerical or structural. The term numerical chromosomal abnormality refers to an abnormal number of chromosomes. Numerical chromosome abnormalities may include chromosome losses or gains. Structural chromosomal abnormalities are alterations in the structure of individual chromosomes, including loss, rearrangement, or gain of chromosome segments. Numerical and structural chromosomal abnormalities may coexist in malignant cells.
Numerical Chromosomal Abnormalities
A cell with a normal complement of chromosomes is called diploid. Cells with 46 chromosomes but with numerical chromosomal abnormalities (e.g., loss of one chromosome and gain of another) are called pseudodiploid. The presence of an abnormal number of chromosomes is called aneuploidy. Cells with more than 46 chromosomes are called hyperdiploid, and those with fewer than 46 chromosomes are called hypodiploid. Loss of one copy of a chromosome results in monosomy for that chromosome; loss of both copies results in nullisomy. Gain of an additional copy of a chromosome results in trisomy of the chromosome that has been gained. Gain of two additional copies of a chromosome results in tetrasomy of that chromosome.
Chromosome loss or gain is indicated in the karyotype by a minus sign or a plus sign followed by the number of the missing or additional chromosome. The number of chromosomes in the cell, given in the karyotype, reflects the chromosomes that have been lost or gained. For example, 45,XY,-7 indicates a male cell with loss of one copy of chromosome 7 (monosomy 7); 47,XX,+8 a female cell with an extra copy of chromosome 8 (trisomy 8); and 48,XY,+13,+13 a male cell with two additional copies of chromosome 13 (tetrasomy 13).
The chromosomes that are most commonly lost in acquired cytogenetic changes include 5, 7, X, and Y (33). Loss of the Y chromosome occurs as an age-related phenomenon in bone marrow cells of normal males (34), but also as an acquired cytogenetic abnormality in malignant cells, either as a sole change (35) or as a secondary change (defined below), particularly in AML with t(8;21) (36). Loss of a chromosome has as its obvious molecular consequence loss of one copy of the genetic material on that chromosome. The genes whose loss is critical in the monosomies that are seen as recurring cytogenetic abnormalities in AML and MDS have not been identified.
The most common acquired trisomy is +8, seen in AML, MDS, and CML in blast crisis (Fig. 3.6) (33). Trisomy 8 occurs both as a sole cytogenetic change and as a secondary cytogenetic abnormality (defined below) in AML. Other trisomies in myeloid disorders include +4, +6, +9, +11, +13, +19, +21, and +22 (33,36,37), whereas the most common trisomies in ALL are +4, +6, +10, +14, +17, +18, +20, +21, and +X (38).
The obvious molecular consequence of a trisomy is the presence of an additional copy of all of the genetic material on a chromosome. Nevertheless, the relationship between trisomies and malignant transformation has remained unclear. Partial tandem duplication of the ALL1 or MLL gene at 11q23 has been demonstrated by molecular techniques in one of the three copies of chromosome 11 in AML with trisomy 11, although all three copies appear structurally normal by conventional cytogenetic analysis (39,40). Exons 2 through 6 or 8 of the ALL1 gene are duplicated, and the partially duplicated ALL1 gene encodes novel partially duplicated RNA and protein species. Demonstration of partial tandem duplication of ALL1 as a consistent finding in AML with trisomy 11 represents the first identification of a specific molecular change associated with a recurrent trisomy in human cancer. Molecular abnormalities have not yet been demonstrated in association with other trisomies. In particular, trisomy 8 has been shown not to be associated with abnormalities in structure or expression of the c-myc oncogene at 8q24 (41).
Numerical chromosomal abnormalities are particularly common in childhood and adult ALL, and their presence is prognostically significant (38,42). Near-haploid or hypodiploid ALL is defined by multiple chromosome losses (43). Hypodiploid ALL most commonly has 45 chromosomes (44). Hyperdiploid ALL is divided into moderate hyperdiploidy, defined by the presence of 47 to 50 chromosomes (45); hyperdiploidy, which is more than 50 chromosomes (46); and near triploidy or near tetraploidy, defined by a markedly increased chromosome number, in the range of 69 to 96 chromosomes (47). Hyperdiploidy and moderate hyperdiploidy are common in childhood ALL, representing 27% and 16% of cases, respectively. Hyperdiploidy is prognostically favorable, whereas moderate hyperdiploidy has an intermediate prognosis (38). Near haploidy and hypodiploidy are prognostically unfavorable (43,44).
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Figure 3.5. Diagrams of normal human G-banded chromosomes showing banding at the 400, 550, and 850 band levels of resolution. (Adapted from ISCN 1995. Basel: Karger, 1995.) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Table 3.1 Common Symbols and Abbreviations Used to Describe Chromosomes and Chromosome Abnormalities |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
Figure 3.6. Trisomy 8 demonstrated by fluorescence in situ hybridization with a chromosome 8 CEP probe. (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) |
FISH with probes hybridizing with the centromeric regions of specific chromosomes allows detection of numerical chromosomal abnormalities in interphase cells (48). FISH may be performed on blood or marrow aspirate smears as well as on bone marrow samples prepared for cytogenetic examination (49), and the presence or absence of numerical chromosomal abnormalities, determined by FISH, may be directly correlated with cell morphology and cell surface antigen expression in blood or marrow smears (50,51).
FISH with a probe specific for the centromere of chromosome 12 is more sensitive than conventional cytogenetic analysis for detecting trisomy 12, the most common cytogenetic abnormality in CLL (52,53). Based on analysis of normal controls, fresh and archived CLL samples are considered to have trisomy 12 by FISH if three chromosome 12 signals are present in more than 4% and 7% of cells, respectively (52,53). In the largest published study of CLL cases, trisomy 12 was present in 35% of cases studied by FISH, but only 6% by conventional cytogenetic analysis (50). Moreover, patients with trisomy 12 detected by FISH had shorter survival times than those without trisomy 12 (53).
Multiple myeloma cells are also difficult to karyotype because of low proliferative rates, even with B-cell mitogens (31), and FISH demonstrates a significantly higher incidence of aneuploidy than is found with conventional cytogenetic analysis (54). Interphase FISH analysis of myeloma cells from 32 patients with probes for chromosomes 1, 3, 7, 8, 11, 12, 16, 17, 18, and X demonstrated aneuploidy in 90% (54). Chromosome gain was more common than chromosome loss, and chromosomes 3, 7, and 11 were most frequently gained. The X chromosome was most frequently lost in females, but not males. Chromosome 17 loss was also common.
Myeloid cells are more easily karyotyped than CLL and myeloma cells, and results of conventional cytogenetic and FISH analysis of AML cells with numerical chromosomal abnormalities such as monosomy 7 and trisomy 8 are more similar than in CLL (55,56). FISH may demonstrate numerical chromosomal abnormalities in cases in which they are not demonstrated by conventional cytogenetic analysis, in which only one metaphase with the abnormality is found by conventional cytogenetic analysis, which is insufficient to define a clone, or in which conventional cytogenetic analysis is inadequate (56).
Structural Chromosomal Abnormalities
A variety of structural chromosomal changes may be present in cells. Precisely defined terms are used to describe these changes. Structural chromosomal abnormalities seen in hematopoietic and lymphoid neoplasms include deletions, isochromosomes, isodicentric chromosomes, inversions, rings, translocations, dicentric chromosomes, additions, insertions, duplications, double-minute chromosomes, and marker chromosomes.
Deletion
A chromosomal deletion (del) is loss of a chromosome segment. Deletions may be either interstitial or terminal. In an interstitial deletion, an internal chromosome segment is lost, and the chromosome segments that are proximal and distal to the deleted segment become juxtaposed. In a terminal deletion, the end of a chromosome arm is deleted. For example, del(5)(q13q33) designates an interstitial deletion of the long arm of chromosome 5 between band q13 and band q33, and del(7)(q22) designates a terminal deletion of chromosome 7 with a break at band q22 and loss of all chromosomal material between band 7q22 and the 7q telomere. Less specific designations such as 7q- or del(7q) are used as abbreviations in text, but should not be used in karyotypes.
Common chromosome deletions include del(5q), del(7q), and del(20q) in AML and MDS; del(13q) in MDS; del(6q) and del(9p) in ALL; and del(13q) in CLL (37). del(7q) is illustrated in Figure 3.7. The critical molecular changes associated with most chromosome deletions are unknown, but loss of tumor suppressor genes is hypothesized (57). The smallest commonly deleted region on 5q is small segment of band 5q31 containing the early growth response 1 protein (EGR1) gene (58) or the interferon regulatory factor-1 (IRF-1) gene (59). A case of AML has been described with deletion of one IRF-1 allele as part of a del(5q), and an inactivating rearrangement of the other allele in the structurally normal chromosome 5 (59). Of note, del(9p) in ALL has been associated with deletion of the interferon genes (60,61). The retinoblastoma gene is deleted in del(13), and the remaining retinoblastoma gene on the cytogenetically normal copy of chromosome 13 may be inactivated (62).
|
|
|
Figure 3.7. G-banded karyotype showing del(7q) (q22q34). (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) |
Isochromosome
An isochromosome (i) is a structurally abnormal chromosome consisting of two identical chromosome arms positioned as mirror images of each other. Isochromosomes may be monocentric (one centromere) or dicentric (two centromeres). A dicentric isochromosome is called an isodicentric chromosome (see below). The most common isochromosomes in hematologic malignancies are i(11q), i(17q), and i(21q) in AML; i(7q), i(9q), and i(17q) in ALL; i(9q), i(17q), and i(22q) in CML; and i(X)(q13), i(17q), and i(21q) in MDS, whereas i(1q), i(6p), i(7q), i(8q), i(9p), i(17q), and i(21q) are recurring isochromosomes in lymphoproliferative disorders and lymphomas (63).
Isochromosome formation leads to both loss and gain of genetic material. As an example, i(17q), which occurs frequently as a secondary cytogenetic abnormality (defined below) in blast crisis of CML and may also occur in ALL (64), consists of two chromosome 17 long arms, without short arms. Cells with i(17q) generally also have one normal chromosome 17; thus, they have one copy of 17p and three copies of 17q. Genetic material on 17p lost in i(17q) formation includes the p53 tumor suppressor gene. Cells with i(17q) have a single copy of p53, which is on the structurally normal chromosome 17. This copy of the p53 gene often has point mutations or other molecular changes that result in p53 inactivation (65,66). i(17q) with inactivating molecular changes in the remaining copy of the p53 gene may also be seen in AML and MDS (67,68).
Isodicentric Chromosome
An isodicentric chromosome (idic) is an isochromosome in which there are two copies of the centromere. Isodicentric chromosomes are rare. An example is idic(7p) in AML (69). idic(7p) consists of two copies of the short arm and the centromere of chromosome 7, positioned as mirror images of each other.
Inversion
A chromosomal inversion (inv) is a structural chromosome change consisting of a 180-degree rotation of a chromosome segment. Inversions may be pericentric or paracentric. In pericentric inversions, the segment that undergoes 180-degree rotation includes the centromere. In paracentric inversions, the inverted segment is entirely within either the short or the long arm of the chromosome, and does not include the centromere. inv(16)(p13q22) is a pericentric inversion (Fig. 3.8), whereas inv(3)(q21q26) is paracentric. Both of these inversions occur in AML (33). Another example of a paracentric inversion is inv(14)(q11q32), which is the most consistent chromosomal change found in T-cell prolymphocytic leukemia (70,71).
The molecular consequence of chromosome inversion is juxtaposition of genes on the long and short arm of a chromosome (pericentric inversion) or on the same arm of a chromosome (paracentric inversion) that are not normally juxtaposed. inv(16)(p13q22), a structural chromosomal abnormality seen in AML (16), is a pericentric inversion that juxtaposes MYHII, the smooth muscle myosin heavy-chain gene at 16p13 with the core binding factor β (CBFβ) gene at 16q22 (72). The reciprocal chromosomal translocation t(16;16)(p13;q22) juxtaposes the same two genes from the two different copies of chromosome 16, rather than the same chromosome 16. inv(3)(q21q26) is a paracentric inversion, also found in AML, that juxtaposes the ribophorin I gene at 3q21 and the EVI I gene at 3q26 (73). Again, the t(3;3)(q21;q26) juxtaposes the same two genes from the two different copies of chromosome 3, rather than the same chromosome 3.
Ring Chromosome
A ring chromosome (r) is an abnormal chromosome in which breaks have occurred in both the short and the long chromosome arms, and the ends have joined together, creating a closed circle, or ring. Ring chromosomes may be derived from one or more chromosomes. They are uncommon in hematologic malignancies and have been reported primarily in AML and CML (33).
|
|
|
Figure 3.8. G-banded karyotype showing inv(16) (p13q22). (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) |
Translocation
A chromosomal translocation is a relocation of material from one chromosome usually to a different chromosome. Translocations are most often reciprocal. A reciprocal translocation is an exchange of material between different chromosomes. Reciprocal translocations generally occur between two chromosomes, but rarely involve more than two chromosomes. Nonreciprocal translocations are rare.
Large numbers of chromosomal translocations have been described in hematopoietic and lymphoid neoplasms. In many instances the associated molecular changes have been identified, and mechanisms of malignant transformation have been elucidated (74). Some translocations result in synthesis of novel fusion proteins. Examples include the t(9;22) in CML, which results in synthesis of an aberrant tyrosine kinase, and a variety of translocations in AML and ALL that result in synthesis of aberrant transcription factors. Alternatively, translocations may result in relocation of an oncogene to a locus that is highly transcribed in a particular cell type, causing activation of the oncogene. Examples include the t(8;14)(q24;q32) in Burkitt lymphoma, the t(8;14) (q24;q11) in T-ALL, and the t(14;18)(q32;q21) in follicular non-Hodgkin lymphomas.
The t(9;22)(q34;q11) in CML, illustrated in Figure 3.9, is a reciprocal translocation between the long arms of chromosomes 9 and 22, with breakpoints at 9q34 and 22q11. The significant molecular change in t(9;22)(q34;q11) is relocation of the abl oncogene from 9q11 to the bcr locus on 22q11 (75,76). A novel bcr/abl fusion gene is created that encodes novel fusion bcr/abl RNA and protein species (77). bcr/abl expression results in increased tyrosine kinase activity, which is sufficient to induce malignant transformation (78,79).
Variant translocations may be seen in CML (80,81,82,83). Simple variant translocations are translocations between 22q11 and a chromosome breakpoint other than 9q34, with bcr/abl fusion at the molecular level. An example is t(17;22)(p13;q11) with bcr/abl rearrangement. Complex variant translocations are three-way translocations between 9q34, 22q11, and a third chromosome breakpoint. An example is t(3;9;22)(q13;q34;q11), which is a three-way translocation between chromosomes 3, 9, and 22 with breaks occurring at 3q13, 9q34, and 22q11, also with bcr/abl fusion.
|
|
|
Figure 3.9. G-banded karyotype showing t(9;22)(q34;q11). A: Fusion (orange/green) signals by fluorescence in situ hybridization demonstrating the t(9;22) and normal untranslocated orange and green signals. B: An extra fusion signal demonstrates presence of extra derivative chromosome 22 (Ph1). (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) |
A number of translocations in acute leukemia result in synthesis of aberrant transcription factors. In the t(8;21) in AML, the AML1 or core binding factor α (CBFα) gene on chromosome 21 is juxtaposed to the ETO gene on chromosome 8 (84,85). The product of the normal CBFα gene dimerizes with the product of the normal CBFβ gene, which is the gene on 16q22 that is involved in inv(16). CBFα and CBFβ form the heterodimeric transcription factor CBF. The AML1/ETO fusion product is an aberrant CBFα protein. Reciprocal translocations with 11q23 breakpoints in AML and ALL also result in synthesis of fusion proteins that are aberrant transcription factors. The gene at 11q23 that is involved in 11q23 translocations is ALL1 or MLL, and reciprocal chromosome breakpoints include 1p32, 1q21, 4q21, 6q27, 9p22, 10p12, 17q21, 19p13.1, and 19p13.3 (86,87,88). t(9;11)(p22;q23) is illustrated in Figure 3.10. In the t(15;17)(q22;q11-12) in APL, illustrated in Figure 3.11A, the retinoic acid receptor α (RARα) gene from chromosome 17 is fused to the PML gene at 15q22. A hybrid PML/RARα gene is created, encoding an aberrant transcription factor that inhibits promyelocyte differentiation (89,90,91,92,93,94).
Examples of translocations resulting in oncogene activation include t(8;14)(q24;q32) in Burkitt lymphoma, t(8;14)(q24;q11) in T-ALL, and t(14;18)(q32;q21) in follicular non-Hodgkin lymphomas. In t(8;14)(q24;q32), the MYC oncogene at 8q24 is juxtaposed to the immunoglobulin heavy-chain gene at 14q32, resulting in dysregulation of MYC transcription (95). In t(8;14)(q24;q11), the MYC oncogene is activated by juxtaposition to the T-cell receptor α/δ locus (96). In t(14;18)(q32;q21), the BCL2 gene at 18q21, which inhibits apoptosis, is overexpressed by virtue of juxtaposition to the immunoglobulin heavy-chain gene at 14q32 (97).
|
|
|
Figure 3.10. A: +8,+8, t(9;11)(p22;q23) G-banded karyotype. (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) B: Chromosomal translocation involving the MLL gene on chromosome 11 (11q23) demonstrated by fluorescence in situ hybridization with a locus-specific probe for MLL. |
Robertsonian translocations are rearrangements that occur between acrocentric chromosomes. They usually occur as constitutional chromosomal abnormalities (98) and are rarely seen as acquired abnormalities associated with malignancy (99).
Several two-color probe systems are available commercially and are used in clinical cytogenetics laboratories for the identification of chromosome translocations by FISH (100,101). FISH may be used to rapidly identify translocations such as t(15;17); a fusion signal is seen, representing juxtaposition of the two probes in the translocation region (Fig. 3.11B). Early confirmation of the diagnosis of APL guides the choice of remission induction therapy. A commercially available locus-specific probe to the MLL gene serves to confirm involvement of this gene in 11q23 chromosome translocations; splitting of the signal is seen (Fig. 3.10B). The chromosomal origins of translocations can also be demonstrated with the use of paint probes (Fig. 3.12).
Derivative Chromosomes
A derivative chromosome (der) is a structurally rearranged chromosome formed by more than one rearrangement within a single chromosome or rearrangements involving two or more chromosomes (7). The term der refers to the chromosome that has the intact centromere. For example, der(22)t(9;22)(q34;q11) means the derivative chromosome 22 has resulted from a translocation of the chromosome 9 segment distal to 9q34 to the long arm of chromosome 22 at band 22q11.
Dicentric Chromosome
A dicentric chromosome (dic) is a structurally abnormal chromosome that has two centromeres. Dicentric chromosomes result from reciprocal translocations in which one of the resulting derivative chromosomes contains the centromeres of both of the chromosomes involved in the translocation. FISH can be helpful in confirming the identity of the centromeres and the other chromosomal material in dicentric chromosomes. Examples of dicentric chromosomes include dic(9;12)(p1?1;p1?2), dic(7;12)(p11;p11), and dic(9;20)(p11;q11.?1) in ALL (102,103,104).
Addition
A chromosomal addition (add) is a gain of chromosomal material of unknown origin attached to a chromosome region or band (7). A plus sign after a chromosome arm is a general designation indicating gain of chromosomal material, resulting in lengthening of the arm. For example, 14q+ indicates the presence of additional chromosomal material on 14q. An abnormal chromosome 14 with extra material on its long arm is a common finding in CLL (30). The abnormal chromosome 14 in CLL can often be identified as resulting from a reciprocal translocation involving the 14q32 breakpoint, such as t(11;14)(q13;q32). However, when a reciprocal translocation is not identified, the designation 14q+ is used to describe an abnormal chromosome 14 with extra chromosomal material of unknown origin on its long arm. Terms such as “14q+” may be used in text to describe these abnormalities but should not be used in karyotypes (7).
Insertion
Insertion (ins) is the presence of a chromosome segment in a new position within the same or another chromosome. Insertions may be direct (dir) or inverted (inv), indicating whether the original orientation is retained or inverted. Insertions are very uncommon. In some instances, structural chromosomal abnormalities that were previously described as insertions have been reinterpreted as translocations. For example, ins(3;3)(q26;q21q26), which describes insertion of the segment of chromosome 3 between bands q21 and q26 into the other chromosome 3 at band 3q26, has been reinterpreted as a reciprocal translocation, t(3;3) (q21;q26) (33).
|
|
|
Figure 3.11. A: G-banded t(15;17) karyotype. B: PML-retinoic acid receptor α (RARα) fusion demonstrated by fluorescence in situ hybridization with locus-specific probes for the PML and RARα genes in an interphase cell. |
Duplication
Duplication (dup) is the presence of an extra copy of a segment of a chromosome, so that there are two copies of the duplicated segment, juxtaposed with one another within the chromosome. Duplications again may be direct (dir) or inverted (inv). An example of a duplication is dup(1)(p12.6q31), which is an uncommon secondary chromosomal abnormality (defined below) in ALL (33). A chromosome duplication results in a structurally abnormal chromosome, whereas a molecular duplication such as the partial tandem duplication of the MLL gene (105) or the FLT3 gene (106,107) is detectable only by molecular techniques.
Double Minutes
A minute (min) is an acentric fragment smaller than the width of a single chromatid. It may be single or double (7). Minutes do not have a banding pattern and occur as a result of gene amplification. Double-minute (dmin) chromosomes are more common in solid tumors than in hematologic or lymphoid malignancies (33). dmin in AML may contain amplified copies of the MYC gene (108), the ALL1 or MLL gene (109), or material from chromosome 19 (110). Homogeneously staining regions (hsr) is the term used to describe abnormally banded or stained regions that contain amplified genes (7).
Marker Chromosomes
The term marker (mar) is used to designate a structurally abnormal chromosome that has no part that can be identified. Karyotypes may include one or more markers. The presence of one marker chromosome in a karyotype is designated by +mar. The presence of several different markers is indicated by +mar1, +mar2, +mar3, and so on. The presence of multiple copies of the same marker is indicated by multiplication signs, as in +mar × 2, +mar × 3, etc. (7). A spectral karyotype of a cell with markers is shown in Figure 3.13.
|
|
|
Figure 3.12. A reciprocal translocation between chromosomes 11 and 20 studied by fluorescence in situ hybridization with whole chromosome paint probes. |
Constitutional versus Acquired Chromosome Changes
Numerical and structural chromosome abnormalities may be either constitutional or acquired. Constitutional chromosome abnormalities are abnormalities that are present in all or almost all cells in the body and exist at the earliest stage of embryogenesis. Acquired chromosomal abnormalities are abnormalities that develop in somatic cells, usually in association with malignant transformation.
The constitutional karyotype of 99% of people is normal (7). Constitutional chromosome abnormalities may be associated with congenital genetic syndromes (such as trisomy 21 with Down syndrome) or may be normal variants that are not associated with somatic changes. The most common constitutional abnormality found in apparently normal people is a pericentric inversion of chromosome 9, inv(9)(p11q13), which is present in approximately 1% of the population. Other constitutional abnormalities include reciprocal translocations, pericentric inversions of chromosomes other than chromosome 9, and robertsonian translocations (111). A constitutional abnormality is designated by c in a karyotype (7). For example, the karyotype of cells from a female with Down syndrome is 47,XX,+21c.
Blood samples are most commonly used to determine constitutional karyotypes. Peripheral blood lymphocytes are stimulated to divide by culturing them with the T-cell mitogen PHA. Blood cells are cultured for 48 or 72 hours in medium with fetal calf serum and PHA. The cells are then processed for cytogenetic analysis using a standard technique. To confirm that an abnormality is constitutional, PHA-stimulated lymphocytes may be studied either in the presence of active disease or in remission. Constitutional abnormalities are also present in a patient’s malignant cells and, unlike acquired abnormalities associated with malignant transformation, remain present when cytogenetic studies are performed following treatment of the malignancy (e.g., when remission bone marrow is studied following treatment of acute leukemia).
Acquired chromosomal abnormalities may develop in cells that previously had a normal karyotype, or may develop as new changes in patients with constitutional chromosome abnormalities. The karyotype 47,XX,+21 designates an acquired trisomy 21 in female cells with a normal constitutional karyotype. In contrast, 47,XX,+21c describes a cell from a female with Down syndrome, without acquired cytogenetic abnormalities. The karyotype 47,XX,t(8;21)(q22;q22),+21c would be that of a cell of a female with Down syndrome with an acquired t(8;21)(q22;q22).
Cytogenetic Definition of Clonality
Once acquired chromosomal abnormalities develop in a cell, the cell and its progeny may have a proliferative or a survival advantage, creating a clone, which is a cell population derived from a single progenitor cell. Cytogenetically, a clone is defined by a minimum of two mitotic cells with gain of the same chromosome or with the same structural abnormality, or three mitotic cells with loss of the same chromosome (112). Cytogenetic changes occurring in an insufficient number of cells to define a clone are considered to be random changes. Alternatively, if apparently random changes can be detected by FISH analysis, FISH may be used to determine their frequency in larger numbers of metaphase spreads and in interphase cells.
Primary versus Secondary Acquired Chromosome Changes
Acquired chromosomal abnormalities may be either primary or secondary. Primary cytogenetic abnormalities are aberrations that are often found as sole chromosomal changes in malignancies. Moreover, they are often associated with specific tumor types. Examples of primary cytogenetic abnormalities include t(8;21) (q22;q22) and inv(16)(p13q22) in AML, t(15;17)(q22;q21) in APL, t(9;22)(q34;q11) in CML, and t(8;14)(q24;q32) in Burkitt lymphoma. Secondary chromosomal abnormalities are changes that generally do not occur by themselves, but rather in addition to primary changes. Secondary changes in acute leukemia include del(5q), del(7q), -7, +8, del(9q), +21, +22, -X, and -Y. Secondary abnormalities associated with blast transformation of CML include +8, i(17q), +der(22), and +19, occurring in addition to t(9;22) (q34;q11).
Primary cytogenetic abnormalities are changes thought to be associated with the pathogenesis of malignant transformation, whereas secondary chromosomal changes likely correlate with tumor progression. Some secondary changes, such as +8, occur in association with a variety of primary changes, while others are associated with specific primary abnormalities, such as del(9q), -X, or -Y with t(8;21). Thus, some secondary changes may confer a proliferative or survival advantage to any cell, while others confer an advantage only to cells with specific primary abnormalities. Trisomy 8 is an example of a cytogenetic change that may be either primary or secondary.
The prognostic significance of secondary chromosome abnormalities is variable. Secondary chromosome abnormalities are present with t(15;17)(q22;q21) in a third of APL patients, and adverse prognostic significance was found in one study (113), but not in two others (114,115). The presence of monosomy 7 as a secondary abnormality further worsens the prognosis of Philadelphia chromosome–positive ALL (116,117), and the presence of secondary chromosome abnormalities, also called clonal evolution, worsens the prognosis of Philadelphia chromosome–positive CML (118,119).
Clinical Applications of Cytogenetics
Diagnosis
The presence of a clonal chromosomal abnormality serves to establish the presence of a clonal bone marrow disorder. Cytogenetic analysis is particularly helpful in establishing the diagnosis of MDS in patients with mild cytopenias and bone marrow cells with minimal or no dysplasia. Similarly, the presence of t(9;22) establishes the diagnosis of CML in patients presenting with slight elevations of the granulocyte count.
|
|
|
Figure 3.13. Spectral karyotype of a cell with markers. (Courtesy of the Clinical Cytogenetics Laboratory, Roswell Park Cancer Institute.) |
Classification
In contrast to the French-American-British classification of hematologic malignancies, the more recent World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues (120) considers cytogenetic analysis to be an essential element of the diagnostic evaluation of neoplastic diseases and incorporates cytogenetic findings into the definition of a number of subtypes. Specifically, presence of t(8;21), inv(16), t(15;17), or 11q23 abnormalities serves to define distinct subsets of AML, and in the presence of t(8;21), inv(16), or t(15;17), the diagnosis of AML is made regardless of the percentage of marrow blasts. Similarly, isolated presence of del(5q) serves to define a distinct subset of MDS (120).
The presence of specific chromosomal abnormalities also serves to define disease entities that are amenable to specific therapies (see below). The prime example is the use of t(15;17) to distinguish APL from other subtypes of AML. In the t(15;17)(q22; q21) in APL, the RARα gene from chromosome 17 is fused to the PML gene at 15q22, creating a hybrid PML/RARα gene encoding an aberrant transcription factor that inhibits promyelocyte differentiation (89,90,91,92,93,94). Classic hypergranular APL is easy to distinguish on the basis of morphology, but hypogranular APL is more difficult to diagnose (121). Both hypergranular and hypogranular APL with t(15;17)(q22;q21) respond to induction therapy with all-trans retinoic acid. The converse problem is the presence of classic APL morphology, but absence of the t(15;17) by conventional cytogenetic analysis (122). Molecular analyses of 60 such patients (122) revealed PML/RARα rearrangements in 42, presence of promyelocytic leukemia zinc finger (PLZF)/RARα rearrangements in 11, and t(5;17) in two; only five had no evidence of RARα rearrangement. In t(11;17)(q23;q21), the RARα gene from chromosome 17 is fused to the PLZF gene at 11q23, rather than to the PML gene at 15q22. The ALL1 or MLL gene at 11q23 is not involved. APL with t(11;17)(q23;q21) rather than t(15;17) is resistant to all-trans retinoic acid induction therapy (123,124).
The t(9;22)(q34;q11) translocation is present in the bone marrow and peripheral blood cells of approximately 95% of patients diagnosed with CML on the basis of their clinical presentation. Patients whose cells do not carry the translocation and in whom the corresponding gene rearrangement is not detected by molecular techniques commonly have atypical features, such as anemia, thrombocytopenia, or massive splenomegaly, and have early disease acceleration and short survival (125,126,127). Cases of CML without the translocation should not be classified as CML, but rather as chronic myelomonocytic leukemias or atypical myeloproliferative syndromes. The presence of t(9;22) demonstrated by cytogenetic, molecular cytogenetic, or molecular biology techniques also provides the data necessary for the decision to treat with imatinib mesylate (Gleevec), the inhibitor of the BCR-ABL tyrosine kinase (128,129).
Etiology
Cytogenetic abnormalities are associated with occupational and environmental exposures in MDS. Notably, exposure to semi-metals and organic chemicals is associated with chromosome 5 and 7 abnormalities, and radiation exposure with chromosome 8 abnormalities (130).
A variety of environmental and occupational exposures have been associated with an increased risk of AML, and cytogenetic abnormalities are frequent in these cases (130,131,132,133,134,135,136,137,138). Cytogenetic abnormalities associated with occupational exposure to organic solvents, petroleum products, and pesticides include -7, del(7q), and del(5q) (132,133,137,139). Smoking is associated with -7, del(7q), -Y, and +13 in AML, and with t(9;22)(q34;q11) in ALL (138).
The incidence of clonal cytogenetic abnormalities in AML developing in patients treated with chemotherapy or radiation therapy for prior malignancies (therapy-related AML) ranges from 75 to 90% in various series (140,141,142). Chromosome 5 or 7 abnormalities such as del(5q), del(7q), or -7 have been found in 72% to 83% of cases of AML in patients with Hodgkin lymphoma, multiple myeloma, ovarian carcinoma, and other malignancies treated with regimens containing the alkylating agents mechlorethamine, melphalan, nitrosoureas, or cyclophosphamide, with or without radiation therapy (142). t(3;21)(q26;q22) has also been associated with alkylating agent therapy (143).
Acute monocytic or myelomonocytic leukemias with cytogenetic abnormalities involving 11q23 are seen in therapy-related leukemia associated with topoisomerase II inhibitors such as anthracyclines and epipodophyllotoxins, rather than alkylating agents (144,145,146). These leukemias develop following treatment for non–small cell and small cell lung carcinoma, ovarian carcinoma, neuroblastoma, osteosarcoma, and ALL (138,144,146). Development of AML with abnormalities of 11q23 is strongly associated with epipodophyllotoxin therapy in patients with T-cell ALL; the cytogenetic findings at the time of development of AML differ from those at earlier presentation of ALL (146). Like most de novo acute leukemias with 11q23 abnormalities, therapy-related acute leukemias with 11q23 abnormalities involve the ALL1 or MLL gene (147,148,149,150).
Rare cases of AML developing in patients treated with chemotherapy for prior malignancies have chromosome abnormalities usually seen in de novo AML, including t(8;21), inv(16), and t(15;17) (151,152). Whereas therapy-related AML with 5, 7, or 11q23 abnormalities has a poor prognosis, therapy-related AML with chromosomal abnormalities typically seen in de novo disease responds well to treatment (151,152).
Prognosis
An extensive literature documents the prognostic significance of recurring chromosomal abnormalities in AML (153,154,155,156,157,158,159,160,161,162,163,164,165), ALL (42,43,44,45,46,51,166,167,168,169,170,171,172,173,174,175), MDS (176,177,178,179), CML (121), CLL (180,181), and NHL (182,183,184,185). Moreover, CML, although defined by a single chromosomal translocation, t(9;22)(q34;q11), may have additional cytogenetic abnormalities that have prognostic significance (119). In addition, deletions in the derivative chromosome 9 in CML, identified by FISH (183), have adverse prognostic significance that may or may not be overcome by imatinib mesylate therapy (184,185). The prognostic significance of chromosome abnormalities in each disease is detailed in the chapters devoted to each of these diseases in this book, and is not reiterated here.
AML, ALL, and MDS cases with normal karyotypes are categorized as having an intermediate response to treatment (159,160,161,170), but treatment response is actually heterogeneous. In AML, recurring molecular abnormalities have been identified that define prognosis of karyotypically normal cases. These include internal tandem duplications of the FLT-3 or MLL gene (105,106,107), expression of the brain and acute leukemia cytoplasmic (BAALC) gene (186), and overexpression of the ETS-related gene (ERG) (187), all of which are unfavorable, and mutations of the CCAAT/enhancer binding protein α (CEPBα) gene (188) and of the nucleophosmin gene (189,190), which are favorable.
Stratification of Treatment
Because of the demonstrated prognostic significance of cytogenetic findings in acute leukemia (38,42,43,44,45,46,47,139,153,154,155,159,160,161,162,163,164,165,166,167,168,169,170,191,192,193,194,195), they are in increasing use in guiding patient management. In particular, they guide the choice of postremission therapy. Patients with AML with the prognostically favorable abnormalities t(8;21) and inv(16) may be managed with postremission chemotherapy rather than transplantation, with the expectation of prolonged disease-free survival and a high cure rate. Conversely, the presence in pretreatment marrow samples of chromosomal abnormalities that predict a low likelihood of long-term disease-free survival with chemotherapy-based treatment strategies is used as a basis for proceeding to bone marrow transplantation in first complete remission CR. Though the chromosomal abnormalities that are prognostically unfavorable with regard to outcome of chemotherapy are also prognostically unfavorable with regard to outcome of transplantation (159,196,197,198), outcome for patients with unfavorable cytogenetics appears to be improved by allogeneic stem cell transplantation (185,196,197). In particular, treatment outcome for Philadelphia chromosome–positive ALL patients is superior following allogeneic bone marrow transplantation, compared to chemotherapy (199,200), though the optimal role of transplantation for Ph-positive ALL in the era of imatinib mesylate therapy is yet unknown.
Given the importance of cytogenetic findings for determining prognosis and for guiding management of acute leukemia, recent studies have sought to compare the sensitivity of molecular cytogenetic and molecular techniques in detecting abnormalities, compared to conventional cytogenetic analysis. Cytogenetics and the reverse transcriptase polymerase chain reaction are almost always concordant in the detection of t(8;21) and inv(16) (201), though the t(8;21)-associated gene rearrangement may be detected in AML with a normal karyotype (202). 11q23 abnormalities do not always involve the MLL gene, but MLL may be rearranged in the absence of 11q23 chromosomal abnormalities (203).
Specific Therapies
The most striking aspect of recent progress in therapy of the hematologic malignancies is the development of specific therapies targeting genetic abnormalities identified at the cytogenetic, molecular cytogenetic, or molecular level. All-trans retinoic acid is an effective agent in the treatment of APL with t(15;17), but APL with t(11;17)(q23;q21) rather than t(15;17) is resistant to all-trans retinoic acid induction therapy (123,124). Imatinib mesylate is a specific inhibitor of the BCR-ABL tyrosine kinase that has activity in Ph-positive CML and ALL (128,129). Imatinib mesylate also inhibits the platelet-derived growth factor (PDGF) and kit kinases and was recently shown to have activity in chronic myeloproliferative disorders with t(5;12)(q33;p13), in which rearrangement of the PDGF receptor β gene on chromosome 5q33 results in its constitutive activation (204). Most recently, imatinib mesylate has been found to have clinical efficacy in hypereosinophilic syndrome (205) and systemic mastocytosis (206) associated with fusion of the PDGFRα (PDGFRα) and Fip1-like 1 (FIP1L1) genes, and deletion of the CHIC2 locus at 4q12, detected by FISH, has been found to serve as a surrogate for FIP1L1-PDGFRA fusion (206) in predicting clinical responsiveness to imatinib mesylate.
Cytogenetic Response
Cytogenetic responses to remittive therapies are strongly predictive of clinical treatment outcome in CML. α-Interferon was the initial therapy found to induce cytogenetic responses in CML (207), albeit in a minority of patients, and to prolong survival in those patients (208). Cytogenetic responses were categorized as complete, if the lowest Ph-positive percentage was 0%; partial, if it was between 1% and 34%; major, if it was below 35% (i.e., major includes complete and partial); and minor, if it was between 35% and 90% (207). These criteria were subsequently applied to defining response to the bcr/abl inhibitor imatinib mesylate, which produces cytogenetic responses in a high proportion of CML patients (209,210,211,212), and has significantly improved survival in this disease. Of interest has been the emergence of new clonal cytogenetic abnormalities in Ph-negative cells in rare patients with CML treated with imatinib mesylate (213,214), with only anecdotal reports of MDS or AML associated with these abnormalities (215).
The most recent treatment response criteria in AML include the category of cytogenetic CR (CRc), indicating reversion to a normal karyotype at CR in AML patients with an abnormal pretreatment karyotype (216). The criteria for defining CRc need to be specified, including the minimum number of metaphases required to define a normal karyotype, and the possible incorporation of FISH to detect pretreatment abnormalities for which there are FISH probes. The importance of CRc was demonstrated in a recent study demonstrating that AML patients with abnormal pretreatment karyotypes who had abnormal, compared to normal, karyotypes at initial morphologic CR following remission induction chemotherapy had significantly shorter disease-free and overall survival and higher cumulative incidence of relapse (217).
Cytogenetic response is also included as a category in the International Working Group (IWG) response criteria in MDS (112). To define the degree of cytogenetic response of cytogenetically abnormal MDS to treatment, analysis of 20 metaphases is optimal, though not mandatory, and FISH may be used to assess changes in the frequency of a specific cytogenetic abnormality. A major cytogenetic response refers to disappearance of a cytogenetic abnormality, and a minor cytogenetic response is 50% or more reduction of abnormal metaphases.
Engraftment
Cytogenetic techniques are used to monitor engraftment of donor cells and persistence or recurrence of host cells following sex-mismatched allogeneic bone marrow transplantation. FISH with probes for the X or Y chromosome allows efficient screening of large numbers of cells with respect to donor or host origin (218).
![]() Conclusion
Conclusion
Cytogenetic analysis has become increasingly sophisticated and has played a progressively larger role in the diagnosis and management of hematologic and lymphoid disorders. This role is expected to continue to expand as a reflection of technical innovations that facilitate cytogenetic analysis, as well as greater knowledge of the clinical, molecular, and therapeutic implications of chromosomal abnormalities.
References
1. Tjio JH, Levan A. The chromosome number of man. Hereditas 1956;42:1–16.
2. Ford CE, Hamerton JL. The chromosomes of man. Nature 1956;178:1020–1023.
3. Lejeune J, Gautier M, Turpin R. Study of somatic chromosomes from 9 mongoloid children. C R Hebd Seances Acad Sci 1959;248:1721–1722.
4. Jacobs PA, Strong JA. A case of human intersexuality having a possible XXY sex-determining mechanism. Nature 1959;183:302–303.
5. Ford CE, Jones KW, Polani PE, et al. A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner’s syndrome). Lancet 1959;1:711–713.
6. Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science 1960;132:1497.
7. ISCN 2005. An international system for human cytogenetic nomenclature. Basel, Switzerland: S Karger, 2004.
8. Caspersson T, Farber S, Foley GE, et al. Chemical differentiation along metaphase chromosomes. Exp Cell Res 1968;49:219–222.
9. Caspersson T, Lomakka G, Zech L. The 24 fluorescence patterns of the human metaphase chromosomes—distinguishing characters and variability. Hereditas 1972;67:89–102.
10. Drets ME, Shaw MW. Specific banding patterns of human chromosomes. Proc Natl Acad Sci U S A 1971;68:2073–2077.
11. Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973;243:290–293.
12. Rowley JD. Identification of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet 1973;16:109–112.
13. Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977;1:549–550.
14. Yunis JJ. High resolution of human chromosomes. Science 1976;191:1268–1270.
15. Yunis JJ, Bloomfield CD, Ensrud K. All patients with acute nonlymphocytic leukemia may have a chromosomal defect. N Engl J Med 1981;305:135–139.
16. Le Beau MM, Larson RA, Bitter MA, et al. Association of an inversion of chromosome 16 with abnormal marrow eosinophils in acute myelomonocytic leukemia. A unique cytogenetic-clinicopathological association. N Engl J Med 1983;309:630–636.
17. Pinkel D, Straume T, Gray JW. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci U S A 1986;83:2934–2938.
18. Cremer T, Landegent J, Bruckner A, et al. Detection of chromosome aberrations in the human interphase nucleus by visualization of specific target DNAs with radioactive and non-radioactive in situ hybridization techniques: diagnosis of trisomy 18 with probe L1.84. Hum Genet 1986;74:346–352.
19. Guan XY, Meltzer PS, Trent JM. Rapid generation of whole chromosome painting probes (WCPs) by chromosome microdissection. Genomics 1994;22:101–107.
20. Speicher MR, Gwyn Ballard S, Ward DC. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet 1996;12:368–375.
21. Schrock E, du Manoir S, Veldman T, et al. Multicolor spectral karyotyping of human chromosomes. Science 1996;273:494–497.
22. Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992;258:818–821.
23. du Manoir S, Speicher MR, Joos S, et al. Detection of complete and partial chromosome gains and losses by comparative genomic in situ hybridization. Hum Genet 1993;90:590–610.
24. Rooney DE, Czepulkowski BH. Human cytogenetics: a practical approach. 2nd ed. New York: Oxford University Press, 1992.
25. Knuutila S, Vuopio P, Elonen E, et al. Culture of bone marrow reveals more cells with chromosomal abnormalities than the direct method in patients with hematologic disorders. Blood 1981;58:369–375.
26. Keinanen M, Knuutila S, Bloomfield CD, et al. The proportion of mitoses in different cell lineages changes during short-term culture of normal human bone marrow. Blood 1986;67:1240–1243.
27. Keinanen M, Knuutila S, Bloomfield CD, et al. Bone marrow cytogenetics: the lineage of dividing cells changes during the first few hours in culture. Leukemia 1987;1:32–37.
28. Larson RA, Kondo K, Vardiman JW, et al. Evidence for a 15;17 translocation in every patient with acute promyelocytic leukemia. Am J Med 1984;76:827–841.
29. Fitzgerald PH, Morris CM, Giles LM. Direct versus cultured preparation of bone marrow cells from 22 patients with acute myeloid leukemia. Hum Genet 1982;60:281–283.
30. Block AW. Cytogenetic analysis of B-cell chronic lymphoproliferative disorders: new technology, complexities and promises. Prog Clin Biol Res 1991;368:145–173.
31. Lai JL, Zandecki M, Mary JY, et al. Improved cytogenetics in multiple myeloma: a study of 151 patients including 117 patients at diagnosis. Blood 1995;85:2490–2497.
32. Hook EB. Exclusion of chromosomal mosaicism: tables of 90%, 95% and 99% confidence limits and comments on use. Am J Hum Genet 1977;29:94–97.
33. Heim S. Cancer cytogenetics. 2nd ed. New York: Wiley, 1995.
34. Loss of the Y chromosome from normal and neoplastic bone marrows. United Kingdom Cancer Cytogenetics Group (UKCCG). Genes Chromosomes Cancer 1992;5:83–88.
35. Riske CB, Morgan R, Ondreyco S, et al. X and Y chromosome loss as sole abnormality in acute non-lymphocytic leukemia (ANLL). Cancer Genet Cytogenet 1994;72:44–47.
36. Slovak ML, Traweek ST, Willman CL, et al. Trisomy 11: an association with stem/progenitor cell immunophenotype. Br J Haematol 1995;90:266–273.
37. Dohner H, Arthur DC, Ball ED, et al. Trisomy 13: a new recurring chromosome abnormality in acute leukemia. Blood 1990;76:1614–1621.
38. Pui CH, Crist WM, Look AT. Biology and clinical significance of cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Blood 1990;76:1449–1463.
39. Caligiuri MA, Strout MP, Schichman SA, et al. Partial tandem duplication of ALL1 as a recurrent molecular defect in acute myeloid leukemia with trisomy 11. Cancer Res 1996;56:1418–1425.
40. Caligiuri MA, Strout MP, Oberkircher AR, et al. The partial tandem duplication of ALL1 in acute myeloid leukemia with normal cytogenetics or trisomy 11 is restricted to one chromosome. Proc Natl Acad Sci U S A 1997;94:3899–3902.
41. Diaz MO, Le Beau MM, Harden A, et al. Trisomy 8 in human hematologic neoplasia and the c-myc and c-mos oncogenes. Leuk Res 1985;9:1437–1442.
42. Cytogenetic abnormalities in adult acute lymphoblastic leukemia: correlations with hematologic findings outcome. A Collaborative Study of the Group Francais de Cytogenetique Hematologique. Blood 1996;87:3135–3142.
43. Pui CH, Carroll AJ, Raimondi SC, et al. Clinical presentation, karyotypic characterization, and treatment outcome of childhood acute lymphoblastic leukemia with a near-haploid or hypodiploid less than 45 line. Blood 1990;75:1170–1177.
44. Pui CH, Williams DL, Raimondi SC, et al. Hypodiploidy is associated with a poor prognosis in childhood acute lymphoblastic leukemia. Blood 1987;70:247–253.
45. Raimondi SC, Roberson PK, Pui CH, et al. Hyperdiploid (47-50) acute lymphoblastic leukemia in children. Blood 1992;79:3245–3252.
46. Pui CH, Raimondi SC, Dodge RK, et al. Prognostic importance of structural chromosomal abnormalities in children with hyperdiploid (greater than 50 chromosomes) acute lymphoblastic leukemia. Blood 1989;73:1963–1967.
47. Pui CH, Carroll AJ, Head D, et al. Near-triploid and near-tetraploid acute lymphoblastic leukemia of childhood. Blood 1990;76:590–596.
48. Anastasi J, Le Beau MM, Vardiman JW, et al. Detection of numerical chromosomal abnormalities in neoplastic hematopoietic cells by in situ hybridization with a chromosome-specific probe. Am J Pathol 1990;136:131–139.
49. Bentz M, Schroder M, Herz M, et al. Detection of trisomy 8 on blood smears using fluorescence in situ hybridization. Leukemia 1993;7:752–757.
50. van Lom K, Hagemeijer A, Smit EM, et al. In situ hybridization on May-Grunwald Giemsa-stained bone marrow and blood smears of patients with hematologic disorders allows detection of cell-lineage-specific cytogenetic abnormalities. Blood 1993;82:884–888.
51. Kibbelaar RE, van Kamp H, Dreef EJ, et al. Combined immunophenotyping and DNA in situ hybridization to study lineage involvement in patients with myelodysplastic syndromes. Blood 1992;79:1823–1828.
52. Anastasi J, Le Beau MM, Vardiman JW, et al. Detection of trisomy 12 in chronic lymphocytic leukemia by fluorescence in situ hybridization to interphase cells: a simple and sensitive method. Blood 1992;79:1796–1801.
53. Escudier SM, Pereira-Leahy JM, Drach JW, et al. Fluorescent in situ hybridization and cytogenetic studies of trisomy 12 in chronic lymphocytic leukemia. Blood 1993;81:2702–2707.
54. Drach J, Schuster J, Nowotny H, et al. Multiple myeloma: high incidence of chromosomal aneuploidy as detected by interphase fluorescence in situ hybridization. Cancer Res 1995;55:3854–3859.
55. Jenkins RB, Le Beau MM, Kraker WJ, et al. Fluorescence in situ hybridization: a sensitive method for trisomy 8 detection in bone marrow specimens. Blood 1992;79:3307–3315.
56. Kibbelaar RE, Mulder JW, Dreef EJ, et al. Detection of monosomy 7 and trisomy 8 in myeloid neoplasia: a comparison of banding and fluorescence in situ hybridization. Blood 1993;82:904–913.
57. Johansson B, Mertens F, Mitelman F. Cytogenetic deletion maps of hematologic neoplasms: circumstantial evidence for tumor suppressor loci. Genes Chromosomes Cancer 1993;8:205–218.
58. Le Beau MM, Espinosa R 3rd, Neuman WL, et al. Cytogenetic and molecular delineation of the smallest commonly deleted region of chromosome 5 in malignant myeloid diseases. Proc Natl Acad Sci U S A 1993;90:5484–5488.
59. Willman CL, Sever CE, Pallavicini MG, et al. Deletion of IRF-1, mapping to chromosome 5q31.1, in human leukemia and preleukemic myelodysplasia. Science 1993;259:968–971.
60. Diaz MO, Rubin CM, Harden A, et al. Deletions of interferon genes in acute lymphoblastic leukemia. N Engl J Med 1990;322:77–82.
61. Einhorn S, Grander D, Bjork O, et al. Deletion of alpha-, beta-, and omega-interferon genes in malignant cells from children with acute lymphocytic leukemia. Cancer Res 1990;50:7781–7785.
62. Hansen MF, Morgan R, Sandberg AA, et al. Structural alterations at the putative retinoblastoma locus in some human leukemias and preleukemia. Cancer Genet Cytogenet 1990;49:15–23.
63. Mertens F, Johansson B, Mitelman F. Isochromosomes in neoplasia. Genes Chromosomes Cancer 1994;10:221–230.
64. Pui CH, Carroll AJ, Raimondi SC, et al. Isochromosomes in childhood acute lymphoblastic leukemia: a collaborative study of 83 cases. Blood 1992;79:2384–2391.
65. Ahuja H, Bar-Eli M, Arlin Z, et al. The spectrum of molecular alterations in the evolution of chronic myelocytic leukemia. J Clin Invest 1991;87:2042–2047.
66. Feinstein E, Cimino G, Gale RP, et al. p53 in chronic myelogenous leukemia in acute phase. Proc Natl Acad Sci U S A 1991;88:6293–6297.
67. Fenaux P, Jonveaux P, Quiquandon I, et al. P53 gene mutations in acute myeloid leukemia with 17p monosomy. Blood 1991;78:1652–1657.
68. Lai JL, Preudhomme C, Zandecki M, et al. Myelodysplastic syndromes and acute myeloid leukemia with 17p deletion. An entity characterized by specific dysgranulopoiesis and a high incidence of P53 mutations. Leukemia 1995;9:370–381.
69. Beverstock GC, de Meijer PH, ten Bokkel Huinink D, et al. A case of isodicentric 7p as sole abnormality in a patient with acute myeloid leukemia. Cancer Genet Cytogenet 1996;89:132–135.
70. Zech L, Godal T, Hammarstrom L, et al. Specific chromosome markers involved with chronic T lymphocyte tumors. Cancer Genet Cytogenet 1986;21:67–77.
71. Matutes E, Brito-Babapulle V, Swansbury J, et al. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood 1991;78:3269–3274.
72. Liu P, Tarle SA, Hajra A, et al. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993;261:1041–1044.
73. Suzukawa K, Parganas E, Gajjar A, et al. Identification of a breakpoint cluster region 3′ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3) (q21q26). Blood 1994;84:2681–2688.
74. Rabbitts TH. Chromosomal translocations in human cancer. Nature 1994;372:143–149.
75. Heisterkamp N, Stephenson JR, Groffen J, et al. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature 1983;306:239–242.
76. Groffen J, Stephenson JR, Heisterkamp N, et al. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984;36:93–99.
77. Shtivelman E, Lifshitz B, Gale RP, et al. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985;315:550–554.
78. Muller AJ, Young JC, Pendergast AM, et al. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol Cell Biol 1991;11:1785–1792.
79. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990;247:824–830.
80. Hagemeijer A, Bartram CR, Smit EM, et al. Is the chromosomal region 9q34 always involved in variants of the Ph1 translocation? Cancer Genet Cytogenet 1984;13:1–16.
81. Morris CM, Rosman I, Archer SA, et al. A cytogenetic and molecular analysis of five variant Philadelphia translocations in chronic myeloid leukemia. Cancer Genet Cytogenet 1988;35:179–197.
82. Dube I, Dixon J, Beckett T, et al. Location of breakpoints within the major breakpoint cluster region (bcr) in 33 patients with bcr rearrangement-positive chronic myeloid leukemia (CML) with complex or absent Philadelphia chromosomes. Genes Chromosomes Cancer 1989;1:106–111.
83. Tosi S, Cabot G, Giudici G, et al. Detection of the breakpoint cluster region-ABL fusion in chronic myeloid leukemia with variant Philadelphia chromosome translocations by in situ hybridization. Cancer Genet Cytogenet 1996;89:153–156.
84. Erickson P, Gao J, Chang KS, et al. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML1/ETO, with similarity to Drosophila segmentation gene, runt. Blood 1992;80:1825–1831.
85. Meyers S, Downing JR, Hiebert SW. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol 1993;13:6336–6345.
86. Cimino G, Nakamura T, Gu Y, et al. An altered 11-kilobase transcript in leukemic cell lines with the t(4;11)(q21;q23) chromosome translocation. Cancer Res 1992;52:3811–3813.
87. Corral J, Forster A, Thompson S, et al. Acute leukemias of different lineages have similar MLL gene fusions encoding related chimeric proteins resulting from chromosomal translocation. Proc Natl Acad Sci U S A 1993;90:8538–8542.
88. Thirman MJ, Gill HJ, Burnett RC, et al. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. N Engl J Med 1993;329:909–914.
89. Alcalay M, Zangrilli D, Pandolfi PP, et al. Translocation breakpoint of acute promyelocytic leukemia lies within the retinoic acid receptor alpha locus. Proc Natl Acad Sci U S A 1991;88:1977–1981.
90. de The H, Chomienne C, Lanotte M, et al. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990;347:558–561.
91. de The H, Lavau C, Marchio A, et al. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991;66:675–684.
92. Kakizuka A, Miller WH Jr, Umesono K, et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991;66:663–674.
93. Longo L, Pandolfi PP, Biondi A, et al. Rearrangements and aberrant expression of the retinoic acid receptor alpha gene in acute promyelocytic leukemias. J Exp Med 1990;172:1571–1575.
94. Grignani F, Ferrucci PF, Testa U, et al. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993;74:423–431.
95. Croce CM, Nowell PC. Molecular basis of human B cell neoplasia. Blood 1985;65:1–7.
96. Erikson J, Finger L, Sun L, et al. Deregulation of c-myc by translocation of the alpha-locus of the T-cell receptor in T-cell leukemias. Science 1986;232:884–886.
97. Lee MS. Molecular aspects of chromosomal translocation t(14;18). Semin Hematol 1993;30:297–305.
98. Tawn EJ, Earl R. The frequencies of constitutional chromosome abnormalities in an apparently normal adult population. Mutat Res 1992;283:69–73.
99. Fan YS, Baer MR, Sait SN, et al. An acquired Robertsonian translocation dic(14;14)(p11;p11) in a patient with a myelodysplastic syndrome following treatment of multiple myeloma. Cancer Genet Cytogenet 1988;30:133–137.
100. Tkachuk DC, Westbrook CA, Andreeff M, et al. Detection of bcr-abl fusion in chronic myelogeneous leukemia by in situ hybridization. Science 1990;250:559–562.
101. Sacchi N, Magnani I, Kearney L, et al. Interphase cytogenetics of the t(8;21)(q22;q22) associated with acute myelogenous leukemia by two-color fluorescence in situ hybridization. Cancer Genet Cytogenet 1995;79:97–103.
102. Huret JL, Heerema NA, Brizard A, et al. Two additional cases of t dic(9:12) in acute lymphocytic leukemia (ALL): prognosis in ALL with dic(9:12). Leukemia 1990;4:423–425.
103. UKCCG. Translocations involving 9p and/or 12p in acute lymphoblastic leukemia. Genes Chromosomes Cancer 1992;5:255–259.
104. Rieder H, Schnittger S, Bodenstein H, et al. dic(9;20): a new recurrent chromosome abnormality in adult acute lymphoblastic leukemia. Genes Chromosomes Cancer 1995;13:54–61.
105. Dohner K, Tobis K, Ulrich R, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol 2002;20:3254–3261.
106. Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002;100:59–66.
107. Frohling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood 2002;100:4372–4380.
108. Slovak ML, Ho JP, Pettenati MJ, et al. Localization of amplified MYC gene sequences to double minute chromosomes in acute myelogenous leukemia. Genes Chromosomes Cancer 1994;9:62–67.
109. Streubel B, Valent P, Jager U, et al. Amplification of the MLL gene on double minutes, a homogeneously staining region, and ring chromosomes in five patients with acute myeloid leukemia or myelodysplastic syndrome. Genes Chromosomes Cancer 2000;27:380–386.
110. Sait SN, Qadir MU, Conroy JM, et al. Double minute chromosomes in acute myeloid leukemia and myelodysplastic syndrome: identification of new amplification regions by fluorescence in situ hybridization and spectral karyotyping. Genes Chromosomes Cancer 2002;34:42–47.
111. Bache I, Hasle H, Tommerup N, et al. Population-based study of cancer among carriers of a constitutional structural chromosomal rearrangement. Genes Chromosomes Cancer 2006;45:231–246.
112. Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006;108:419–425.
113. Hiorns LR, Swansbury GJ, Mehta J, et al. Additional chromosome abnormalities confer worse prognosis in acute promyelocytic leukaemia. Br J Haematol 1997;96:314–321.
114. Schoch C, Haase D, Haferlach T, et al. Incidence and implication of additional chromosome aberrations in acute promyelocytic leukaemia with translocation t(15;17)(q22;q21): a report on 50 patients. Br J Haematol 1996;94:493–500.
115. Slack JL, Arthur DC, Lawrence D, et al. Secondary cytogenetic changes in acute promyelocytic leukemia–prognostic importance in patients treated with chemotherapy alone and association with the intron 3 breakpoint of the PML gene: a Cancer and Leukemia Group B study. J Clin Oncol 1997;15:1786–1795.
116. Rieder H, Ludwig WD, Gassmann W, et al. Prognostic significance of additional chromosome abnormalities in adult patients with Philadelphia chromosome positive acute lymphoblastic leukaemia. Br J Haematol 1996;95:678–691.
117. Russo C, Carroll A, Kohler S, et al. Philadelphia chromosome and monosomy 7 in childhood acute lymphoblastic leukemia: a Pediatric Oncology Group study. Blood 1991;77:1050–1056.
118. Sokal JE, Gomez GA, Baccarani M, et al. Prognostic significance of additional cytogenetic abnormalities at diagnosis of Philadelphia chromosome-positive chronic granulocytic leukemia. Blood 1988;72:294–298.
119. Cortes JE, Talpaz M, Giles F, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood 2003;101:3794–3800.
120. Jaffe ES, Harris NL, Stein H, et al. World Health Organization classification of tumours, pathology & genetics of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press, 2001.
121. Golomb HM, Rowley JD, Vardiman JW, et al. “Microgranular” acute promyelocytic leukemia: a distinct clinical, ultrastructural, and cytogenetic entity. Blood 1980;55:253–259.
122. Grimwade D, Biondi A, Mozziconacci MJ, et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Francais de Cytogenetique Hematologique, Groupe de Francais d’Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action “Molecular Cytogenetic Diagnosis in Haematological Malignancies”. Blood 2000;96:1297–1308.
123. Guidez F, Huang W, Tong JH, et al. Poor response to all-trans retinoic acid therapy in a t(11;17) PLZF/RAR alpha patient. Leukemia 1994;8:312–317.
124. Licht JD, Chomienne C, Goy A, et al. Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 1995;85:1083–1094.
125. Fitzgerald PH, Beard ME, Morris CM, et al. Ph-negative chronic myeloid leukaemia. Br J Haematol 1987;66:311–314.
126. Kurzrock R, Kantarjian HM, Shtalrid M, et al. Philadelphia chromosome-negative chronic myelogenous leukemia without breakpoint cluster region rearrangement: a chronic myeloid leukemia with a distinct clinical course. Blood 1990;75:445–452.
127. Dobrovic A, Morley AA, Seshadri R, et al. Molecular diagnosis of Philadelphia negative CML using the polymerase chain reaction and DNA analysis: clinical features and course of M-bcr negative and M-bcr positive CML. Leukemia 1991;5:187–190.
128. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001;344:1038–1042.
129. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031–1037.
130. West RR, Stafford DA, White AD, et al. Cytogenetic abnormalities in the myelodysplastic syndromes and occupational or environmental exposure. Blood 2000;95:2093–2097.
131. Mitelman F, Nilsson PG, Brandt L, et al. Chromosome pattern, occupation, and clinical features in patients with acute nonlymphocytic leukemia. Cancer Genet Cytogenet 1981;4:197–214.
132. Mitelman F, Brandt L, Nilsson PG. Relation among occupational exposure to potential mutagenic/carcinogenic agents, clinical findings, and bone marrow chromosomes in acute nonlymphocytic leukemia. Blood 1978;52:1229–1237.
133. Golomb HM, Alimena G, Rowley JD, et al. Correlation of occupation and karyotype in adults with acute nonlymphocytic leukemia. Blood 1982;60:404–411.
134. Mitelman F, Nilsson PG, Brandt L. Fourth International Workshop on Chromosomes in Leukemia 1982: correlation of karyotype and occupational exposure to potential mutagenic/carcinogenic agents in acute nonlymphocytic leukemia. Cancer Genet Cytogenet 1984;11:326–331.
135. Sandler DP, Collman GW. Cytogenetic and environmental factors in the etiology of the acute leukemias in adults. Am J Epidemiol 1987;126:1017–1032.
136. Crane MM, Keating MJ, Trujillo JM, et al. Environmental exposures in cytogenetically defined subsets of acute nonlymphocytic leukemia. JAMA 1989;262:634–639.
137. Cuneo A, Fagioli F, Pazzi I, et al. Morphologic, immunologic and cytogenetic studies in acute myeloid leukemia following occupational exposure to pesticides and organic solvents. Leuk Res 1992;16:789–796.
138. Sandler DP, Shore DL, Anderson JR, et al. Cigarette smoking and risk of acute leukemia: associations with morphology and cytogenetic abnormalities in bone marrow. J Natl Cancer Inst 1993;85:1994–2003.
139. Bloomfield CD, Goldman A, Hassfeld D, et al. Fourth International Workshop on Chromosomes in Leukemia 1982: clinical significance of chromosomal abnormalities in acute nonlymphoblastic leukemia. Cancer Genet Cytogenet 1984;11:332–350.
140. Bloomfield CD. Chromosome abnormalities in secondary myelodysplastic syndromes. Scand J Haematol Suppl 1986;45:82–90.
141. Kantarjian HM, Keating MJ, Walters RS, et al. Therapy-related leukemia and myelodysplastic syndrome: clinical, cytogenetic, and prognostic features. J Clin Oncol 1986;4:1748–1757.
142. Pedersen-Bjergaard J, Philip P, Larsen SO, et al. Chromosome aberrations and prognostic factors in therapy-related myelodysplasia and acute nonlymphocytic leukemia. Blood 1990;76:1083–1091.
143. Rubin CM, Larson RA, Anastasi J, et al. t(3;21)(q26;q22): a recurring chromosomal abnormality in therapy-related myelodysplastic syndrome and acute myeloid leukemia. Blood 1990;76:2594–2598.
144. Pedersen-Bjergaard J, Philip P, Ravn V, et al. Therapy-related acute nonlymphocytic leukemia of FAB type M4 or M5 with early onset and t(9;11) (p21;q23) or a normal karyotype: a separate entity? J Clin Oncol 1988;6:395–397.
145. Ratain MJ, Kaminer LS, Bitran JD, et al. Acute nonlymphocytic leukemia following etoposide and cisplatin combination chemotherapy for advanced non-small-cell carcinoma of the lung. Blood 1987;70:1412–1417.
146. Pui CH, Behm FG, Raimondi SC, et al. Secondary acute myeloid leukemia in children treated for acute lymphoid leukemia. N Engl J Med 1989;321:136–142.
147. Super HJ, McCabe NR, Thirman MJ, et al. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 1993;82:3705–3711.
148. Hunger SP, Tkachuk DC, Amylon MD, et al. HRX involvement in de novo and secondary leukemias with diverse chromosome 11q23 abnormalities. Blood 1993;81:3197–3203.
149. Felix CA, Hosler MR, Winick NJ, et al. ALL-1 gene rearrangements in DNA topoisomerase II inhibitor-related leukemia in children. Blood 1995;85:3250–3256.
150. Domer PH, Head DR, Renganathan N, et al. Molecular analysis of 13 cases of MLL/11q23 secondary acute leukemia and identification of topoisomerase II consensus-binding sequences near the chromosomal breakpoint of a secondary leukemia with the t(4;11). Leukemia 1995;9:1305–1312.
151. Fenaux P, Lucidarme D, Lai JL, et al. Favorable cytogenetic abnormalities in secondary leukemia. Cancer 1989;63:2505–2508.
152. Quesnel B, Kantarjian H, Bjergaard JP, et al. Therapy-related acute myeloid leukemia with t(8;21), inv(16), and t(8;16): a report on 25 cases and review of the literature. J Clin Oncol 1993;11:2370–2379.
153. Fenaux P, Preudhomme C, Lai JL, et al. Cytogenetics and their prognostic value in de novo acute myeloid leukaemia: a report on 283 cases. Br J Haematol 1989;73:61–67.
154. Arthur DC, Berger R, Golomb HM, et al. The clinical significance of karyotype in acute myelogenous leukemia. Cancer Genet Cytogenet 1989;40:203–216.
155. Schiffer CA, Lee EJ, Tomiyasu T, et al. Prognostic impact of cytogenetic abnormalities in patients with de novo acute nonlymphocytic leukemia. Blood 1989;73:263–270.
156. Dastugue N, Payen C, Lafage-Pochitaloff M, et al. Prognostic significance of karyotype in de novo adult acute myeloid leukemia. The BGMT group. Leukemia 1995;9:1491–1498.
157. Kalwinsky DK, Raimondi SC, Schell MJ, et al. Prognostic importance of cytogenetic subgroups in de novo pediatric acute nonlymphocytic leukemia. J Clin Oncol 1990;8:75–83.
158. Martinez-Climent JA, Lane NJ, Rubin CM, et al. Clinical and prognostic significance of chromosomal abnormalities in childhood acute myeloid leukemia de novo. Leukemia 1995;9:95–101.
159. Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 2000;96:4075–4083.
160. Grimwade D, Walker H, Harrison G, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001;98:1312–1320.
161. Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002;100:4325–4336.
162. Bloomfield CD, Secker-Walker LM, Goldman AI, et al. Six-year follow-up of the clinical significance of karyotype in acute lymphoblastic leukemia. Cancer Genet Cytogenet 1989;40:171–185.
163. Crist W, Carroll A, Shuster J, et al. Philadelphia chromosome positive childhood acute lymphoblastic leukemia: clinical and cytogenetic characteristics and treatment outcome. A Pediatric Oncology Group study. Blood 1990;76:489–494.
164. Fletcher JA, Lynch EA, Kimball VM, et al. Translocation (9;22) is associated with extremely poor prognosis in intensively treated children with acute lymphoblastic leukemia. Blood 1991;77:435–439.
165. Secker-Walker LM, Craig JM, Hawkins JM, et al. Philadelphia positive acute lymphoblastic leukemia in adults: age distribution, BCR breakpoint and prognostic significance. Leukemia 1991;5:196–199.
166. Preti HA, O’Brien S, Giralt S, et al. Philadelphia-chromosome-positive adult acute lymphocytic leukemia: characteristics, treatment results, and prognosis in 41 patients. Am J Med 1994;97:60–65.
167. Raimondi SC, Behm FG, Roberson PK, et al. Cytogenetics of pre-B-cell acute lymphoblastic leukemia with emphasis on prognostic implications of the t(1;19). J Clin Oncol 1990;8:1380–1388.
168. Crist WM, Carroll AJ, Shuster JJ, et al. Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study. Blood 1990;76:117–122.
169. Pui CH, Frankel LS, Carroll AJ, et al. Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia with the t(4;11)(q21;q23): a collaborative study of 40 cases. Blood 1991;77:440–447.
170. Wetzler M, Dodge RK, Mrozek K, et al. Prospective karyotype analysis in adult acute lymphoblastic leukemia: the cancer and leukemia Group B experience. Blood 1999;93:3983–3993.
171. Yunis JJ, Rydell RE, Oken MM, et al. Refined chromosome analysis as an independent prognostic indicator in de novo myelodysplastic syndromes. Blood 1986;67:1721–1730.
172. Billstrom R, Thiede T, Hansen S, et al. Bone marrow karyotype and prognosis in primary myelodysplastic syndromes. Eur J Haematol 1988;41:341–346.
173. Morel P, Hebbar M, Lai JL, et al. Cytogenetic analysis has strong independent prognostic value in de novo myelodysplastic syndromes and can be incorporated in a new scoring system: a report on 408 cases. Leukemia 1993;7:1315–1323.
174. Toyama K, Ohyashiki K, Yoshida Y, et al. Clinical implications of chromosomal abnormalities in 401 patients with myelodysplastic syndromes: a multicentric study in Japan. Leukemia 1993;7:499–508.
175. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079–2088.
176. Han T, Henderson ES, Emrich LJ, et al. Prognostic significance of karyotypic abnormalities in B cell chronic lymphocytic leukemia: an update. Semin Hematol 1987;24:257–263.
177. Juliusson G, Oscier DG, Fitchett M, et al. Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med 1990;323:720–724.
178. Byrd JC, Smith L, Hackbarth ML, et al. Interphase cytogenetic abnormalities in chronic lymphocytic leukemia may predict response to rituximab. Cancer Res 2003;63:36–38.
179. Kristoffersson U, Heim S, Mandahl N, et al. Prognostic implication of cytogenetic findings in 106 patients with non-Hodgkin lymphoma. Cancer Genet Cytogenet 1987;25:55–64.
180. Levine EG, Arthur DC, Frizzera G, et al. Cytogenetic abnormalities predict clinical outcome in non-Hodgkin lymphoma. Ann Intern Med 1988;108:14–20.
181. Schouten HC, Sanger WG, Weisenburger DD, et al. Chromosomal abnormalities in untreated patients with non-Hodgkin’s lymphoma: associations with histology, clinical characteristics, and treatment outcome. The Nebraska Lymphoma Study Group. Blood 1990;75:1841–1847.
182. Offit K, Wong G, Filippa DA, et al. Cytogenetic analysis of 434 consecutively ascertained specimens of non-Hodgkin’s lymphoma: clinical correlations. Blood 1991;77:1508–1515.
183. Huntly BJ, Reid AG, Bench AJ, et al. Deletions of the derivative chromosome 9 occur at the time of the Philadelphia translocation and provide a powerful and independent prognostic indicator in chronic myeloid leukemia. Blood 2001;98:1732–1738.
184. Huntly BJ, Guilhot F, Reid AG, et al. Imatinib improves but may not fully reverse the poor prognosis of patients with CML with derivative chromosome 9 deletions. Blood 2003;102:2205–2212.
185. Quintas-Cardama A, Kantarjian H, Talpaz M, et al. Imatinib mesylate therapy may overcome the poor prognostic significance of deletions of derivative chromosome 9 in patients with chronic myelogenous leukemia. Blood 2005;105:2281–2286.
186. Baldus CD, Tanner SM, Ruppert AS, et al. BAALC expression predicts clinical outcome of de novo acute myeloid leukemia patients with normal cytogenetics: a Cancer and Leukemia Group B Study. Blood 2003;102:1613–1618.
187. Marcucci G, Baldus CD, Ruppert AS, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol 2005;23:9234–9242.
188. Frohling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol 2004;22:624–633.
189. Schnittger S, Schoch C, Kern W, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 2005;106:3733–3739.
190. Thiede C, Koch S, Creutzig E, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 2006;107:4011–4020.
191. Berger R, Bernheim A, Ochoa-Noguera ME, et al. Prognostic significance of chromosomal abnormalities in acute nonlymphocytic leukemia: a study of 343 patients. Cancer Genet Cytogenet 1987;28:293–299.
192. Keating MJ, Smith TL, Kantarjian H, et al. Cytogenetic pattern in acute myelogenous leukemia: a major reproducible determinant of outcome. Leukemia 1988;2:403–412.
193. Samuels BL, Larson RA, Le Beau MM, et al. Specific chromosomal abnormalities in acute nonlymphocytic leukemia correlate with drug susceptibility in vivo. Leukemia 1988;2:79–83.
194. Yunis JJ, Lobell M, Arnesen MA, et al. Refined chromosome study helps define prognostic subgroups in most patients with primary myelodysplastic syndrome and acute myelogenous leukaemia. Br J Haematol 1988;68:189–194.
195. Westbrook CA, Hooberman AL, Spino C, et al. Clinical significance of the BCR-ABL fusion gene in adult acute lymphoblastic leukemia: a Cancer and Leukemia Group B Study (8762). Blood 1992;80:2983–2990.
196. Ferrant A, Doyen C, Delannoy A, et al. Karyotype in acute myeloblastic leukemia: prognostic significance in a prospective study assessing bone marrow transplantation in first remission. Bone Marrow Transplant 1995;15:685–690.
197. Gale RP, Horowitz MM, Weiner RS, et al. Impact of cytogenetic abnormalities on outcome of bone marrow transplants in acute myelogenous leukemia in first remission. Bone Marrow Transplant 1995;16:203–208.
198. Slovak ML, Kopecky KJ, Wolman SR, et al. Cytogenetic correlation with disease status and treatment outcome in advanced stage leukemia post bone marrow transplantation: a Southwest Oncology Group study (SWOG-8612). Leuk Res 1995;19:381–388.
199. Forman SJ, O’Donnell MR, Nademanee AP, et al. Bone marrow transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 1987;70:587–588.
200. Barrett AJ, Horowitz MM, Ash RC, et al. Bone marrow transplantation for Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 1992;79:3067–3070.
201. Mrozek K, Prior TW, Edwards C, et al. Comparison of cytogenetic and molecular genetic detection of t(8;21) and inv(16) in a prospective series of adults with de novo acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol 2001;19:2482–2492.
202. Sarriera JE, Albitar M, Estrov Z, et al. Comparison of outcome in acute myelogenous leukemia patients with translocation (8;21) found by standard cytogenetic analysis and patients with AML1/ETO fusion transcript found only by PCR testing. Leukemia 2001;15:57–61.
203. Ibrahim S, Estey EH, Pierce S, et al. 11q23 abnormalities in patients with acute myelogenous leukemia and myelodysplastic syndrome as detected by molecular and cytogenetic analyses. Am J Clin Pathol 2000;114:793–797.
204. Apperley JF, Gardembas M, Melo JV, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med 2002;347:481–487.
205. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003;348:1201–1214.
206. Pardanani A, Ketterling RP, Brockman SR, et al. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood 2003;102:3093–3096.
207. Talpaz M, Kantarjian HM, McCredie K, et al. Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha A in chronic myelogenous leukemia. N Engl J Med 1986;314:1065–1069.
208. Kantarjian HM, Smith TL, O’Brien S, et al. Prolonged survival in chronic myelogenous leukemia after cytogenetic response to interferon-alpha therapy. The Leukemia Service. Ann Intern Med 1995;122:254–261.
209. Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood 2002;99:1928–1937.
210. Kantarjian HM, Cortes JE, O’Brien S, et al. Imatinib mesylate therapy in newly diagnosed patients with Philadelphia chromosome-positive chronic myelogenous leukemia: high incidence of early complete and major cytogenetic responses. Blood 2003;101:97–100.
211. Kantarjian HM, Cortes JE, O’Brien S, et al. Long-term survival benefit and improved complete cytogenetic and molecular response rates with imatinib mesylate in Philadelphia chromosome-positive chronic-phase chronic myeloid leukemia after failure of interferon-alpha. Blood 2004;104:1979–1988.
212. O’Dwyer ME, Mauro MJ, Blasdel C, et al. Clonal evolution and lack of cytogenetic response are adverse prognostic factors for hematologic relapse of chronic phase CML patients treated with imatinib mesylate. Blood 2004;103:451–455.
213. Bumm T, Muller C, Al-Ali HK, et al. Emergence of clonal cytogenetic abnormalities in Ph- cells in some CML patients in cytogenetic remission to imatinib but restoration of polyclonal hematopoiesis in the majority. Blood 2003;101:1941–1949.
214. Terre C, Eclache V, Rousselot P, et al. Report of 34 patients with clonal chromosomal abnormalities in Philadelphia-negative cells during imatinib treatment of Philadelphia-positive chronic myeloid leukemia. Leukemia 2004;18:1340–1346.
215. Kovitz C, Kantarjian H, Garcia-Manero G, et al. Myelodysplastic syndromes and acute leukemia developing after imatinib mesylate therapy for chronic myeloid leukemia. Blood 2006;108:2811–2813.
216. Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003;21:4642–4649.
217. Marcucci G, Mrozek K, Ruppert AS, et al. Abnormal cytogenetics at date of morphologic complete remission predicts short overall and disease-free survival, and higher relapse rate in adult acute myeloid leukemia: results from cancer and leukemia group B study 8461. J Clin Oncol 2004;22:2410–2418.
218. Wessman M, Ruutu T, Volin L, et al. In situ hybridization using a Y-specific probe—a sensitive method for distinguishing residual male recipient cells from female donor cells in bone marrow transplantation. Bone Marrow Transplant 1989;4:283–286.