CHAPTER CONTENTS
Introduction
Type I: Immediate (Anaphylactic) Hypersensitivity
Atopy
Drug Hypersensitivity
Desensitization
Treatment & Prevention
Type II: Cytotoxic Hypersensitivity
Type III: Immune Complex Hypersensitivity
Arthus Reaction
Serum Sickness
Immune Complex Diseases
Type IV: Delayed (Cell-Mediated) Hypersensitivity
Clinically Important Delayed Hypersensitivity Reactions
Self-Assessment Questions
Practice Questions: USMLE & Course Examinations
INTRODUCTION
Hypersensitivity is the term used when an immune response results in exaggerated or inappropriate reactions harmful to the host. The term allergy is often equated with hypersensitivity but more accurately should be limited to the IgE–mediated reactions discussed later in the section “Type I: Immediate (Anaphylactic) Hypersensitivity.”
The clinical manifestations of these reactions are typical in a given individual and occur on contact with the specific antigen to which the individual is hypersensitive. The first contact of the individual with the antigen sensitizes (i.e., induces the antibody), and the subsequent contacts elicit the allergic response.
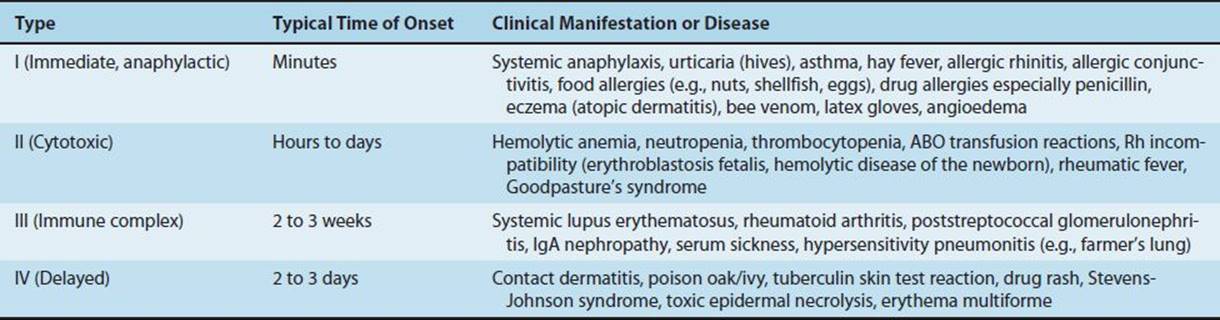
Hypersensitivity reactions can be subdivided into four main types. Types I, II, and III are antibody-mediated, whereas type IV is cell-mediated (Table 65–1). Type I reactions are mediated by IgE, whereas types II and III are mediated by IgG. The immunologic reactions are summarized in Table 65–1. The clinical manifestations of the hypersensitivity reactions are described in Table 65–2.
TABLE 65–1 Immunologic Aspects of Hypersensitivity Reactions
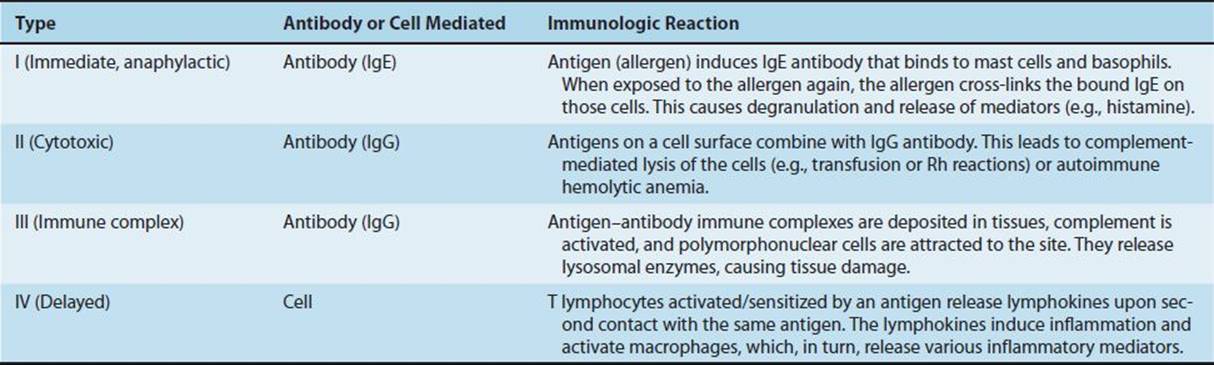
TABLE 65–2 Clinical Manifestations of Hypersensitivity Reactions
TYPE I: IMMEDIATE (ANAPHYLACTIC) HYPERSENSITIVITY
An immediate hypersensitivity reaction occurs when an antigen (allergen) binds to IgE on the surface of mast cells with the consequent release of several mediators (see list of mediators that follows) (Figure 65–1). The process begins when an antigen induces the formation of IgE antibody, which binds firmly by its Fc portion to receptors on the surface of basophils and mast cells. Reexposure to the same antigen results in cross-linking of the cell-bound IgE, degranulation, and release of pharmacologically active mediators within minutes (immediate phase). Cyclic nucleotides and calcium play essential roles in release of the mediators.1 Symptoms such as edema and erythema (“wheal and flare”) and itching appear rapidly because these mediators (e.g., histamine) are preformed.
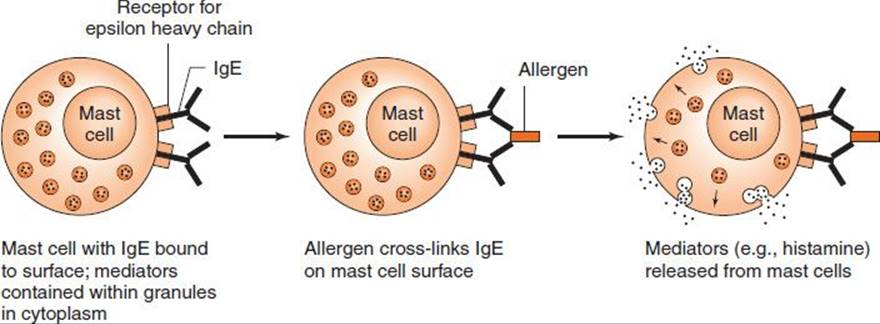
FIGURE 65–1 Immediate (anaphylactic) hypersensitivity.
The late phase of IgE-mediated inflammation occurs approximately 6 hours after exposure to the antigen and is due to mediators (e.g., leukotrienes [SRS-A]) that are synthesized after the cell degranulates. These mediators cause an influx of inflammatory cells, such as neutrophils and eosinophils, and symptoms such as erythema and induration occur. For example, eosinophils play a major role in the late-phase reaction in asthma.
Complement is not involved with either the immediate or late reactions because IgE does not activate complement.
Note that the allergens involved in hypersensitivity reactions are substances, such as pollens, animal danders, foods (nuts, shellfish), and various drugs, to which most people do not exhibit clinical symptoms. However, some individuals respond to those substances by producing large amounts of IgE and, as a result, manifest various allergic symptoms. The increased IgE is the result of increased class switching to IgE in B cells caused by large amounts of interleukin (IL)-4 produced by Th-2 cells. Nonallergic individuals respond to the same antigen by producing IgG, which does not cause the release of mediators from mast cells and basophils. (There are no receptors for IgG on those cells.) There is a genetic predisposition to immediate hypersensitivity reactions, which is discussed in the “Atopy” section later.
The clinical manifestations of type I hypersensitivity can appear in various forms (e.g., urticaria [also known as hives], eczema, rhinitis and conjunctivitis [also known as hay fever], and asthma). Which clinical manifestation occurs depends in large part on the route of entry of the allergen and on the location of the mast cells bearing the IgE specific for the allergen. For example, some individuals exposed to pollens in the air get hay fever, whereas others who ingest allergens in food get diarrhea. Furthermore, people who respond to an allergen with urticaria have the allergen-specific IgE on mast cells in the skin, whereas those who respond with rhinitis have the allergen-specific mast cells in the nose.
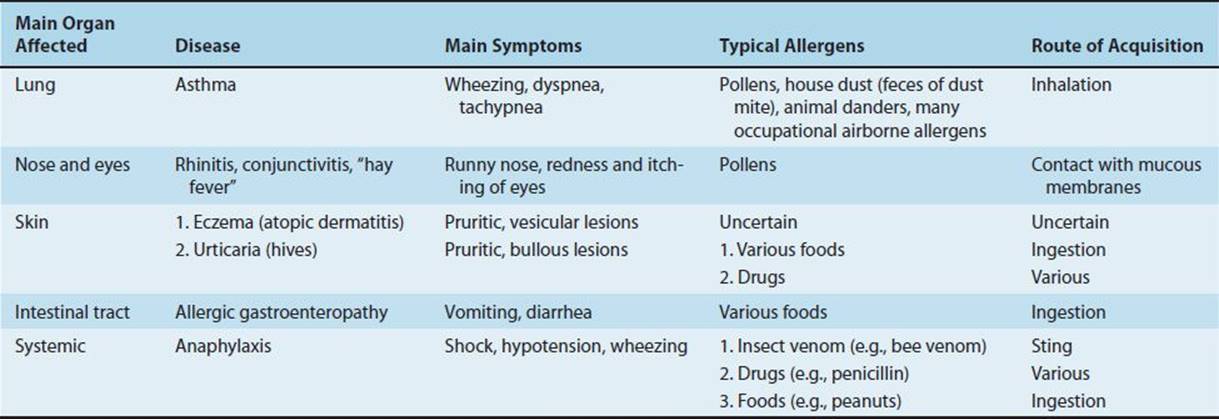
The most severe form of type I hypersensitivity is systemic anaphylaxis, in which severe bronchoconstriction and hypotension (shock) can be life-threatening. The most common causes of anaphylaxis are foods such as peanuts and shellfish, bee venom, and drugs such as penicillin. Of particular interest to medical personnel are type I hypersensitivity reactions to the wearing of latex rubber gloves, which include urticaria, asthma, and even systemic anaphylaxis. Table 65–3 summarizes some of the important clinical aspects of immediate hypersensitivities.
TABLE 65–3 Important Clinical Aspects of Immediate Hypersensitivities
No single mediator accounts for all the manifestations of type I hypersensitivity reactions. Some important mediators and their effects are as follows:
(1) Histamine occurs in granules of tissue mast cells and basophils in a preformed state. Its release causes vasodilation, increased capillary permeability, and smooth muscle contraction. Clinically, disorders such as allergic rhinitis (hay fever), urticaria, and angioedema can occur. The bronchospasm so prominent in acute anaphylaxis results, in part, from histamine release. Antihistamine drugs block histamine receptor sites and can be relatively effective in allergic rhinitis but not in asthma (see later).
(2) Slow-reacting substance of anaphylaxis (SRS-A) consists of several leukotrienes, which do not exist in a preformed state but are produced during anaphylactic reactions. This accounts for the slow onset of the effect of SRS-A. Leukotrienes are formed from arachidonic acid by the lipoxygenase pathway and cause increased vascular permeability and smooth muscle contraction. They are the principal mediators in the bronchoconstriction of asthma and are not influenced by antihistamines.
(3) Eosinophil chemotactic factor of anaphylaxis (ECF-A) is a tetrapeptide that exists preformed in mast cell granules. When released during anaphylaxis, it attracts eosinophils that are prominent in immediate allergic reactions. The role of eosinophils in type I hypersensitivity reactions is uncertain, but they do release histaminase and arylsulfatase, which degrade two important mediators, histamine and SRS-A, respectively. Eosinophils may therefore reduce the severity of the type I response.
(4) Serotonin (hydroxytryptamine) is preformed in mast cells and blood platelets. When released during anaphylaxis, it causes capillary dilation, increased vascular permeability, and smooth muscle contraction but is of minor importance in human anaphylaxis.
(5) Prostaglandins and thromboxanes are related to leukotrienes. They are derived from arachidonic acid via the cyclooxygenase pathway. Prostaglandins cause dilation and increased permeability of capillaries and bronchoconstriction. Thromboxanes aggregate platelets.
(6) Platelet-activating factor (PAF) is a phospholipid produced by mast cells that can cause bronchoconstriction, hypotension, and vascular permeability.
The aforementioned mediators are active only for a few minutes after release; they are enzymatically inactivated and resynthesized slowly. Manifestations of anaphylaxis vary among species because mediators are released at different rates in different amounts, and tissues vary in their sensitivity to them. For example, the respiratory tract (bronchospasm, laryngeal edema) is a principal shock organ in humans, but the liver (hepatic veins) plays that role in dogs.
In allergic airway disease (asthma), the airway hyperactivity appears to be caused by IL-13. IL-13 is made by Th-2 cells and binds to a receptor that shares a chain with the IL-4 receptor. IL-13 does not increase the amount of IgE. Lebrikizumab, a monoclonal antibody against IL-13, reduces symptoms in some patients with severe asthma.
In contrast to anaphylactic reactions, which are IgE-mediated, anaphylactoid reactions, which appear clinically similar to anaphylactic ones, are not IgE-mediated. In anaphylactoid reactions, the inciting agents, usually drugs or iodinated contrast media, directly induce the mast cells and basophils to release their mediators without the involvement of IgE.
Atopy
Atopic disorders, such as hay fever, asthma, eczema, and urticaria, are immediate-hypersensitivity reactions that exhibit a strong familial predisposition and are associated with elevated IgE levels. Several processes seem likely to play a role in atopy, for example, failure of regulation at the T-cell level (e.g., increased production of IL-4 leads to increased IgE synthesis), enhanced uptake and presentation of environmental antigens, and hyperreactivity of target tissues. Target tissues often contain large numbers of Th-2 cells, and these are thought to play a major role in the pathogenesis of atopic reactions.
It is estimated that up to 40% of people in the United States have experienced an atopic disorder at some time in their lives. The incidence of allergic diseases, such as asthma, is increasing markedly in the developed countries of North America and Europe. One hypothesis that might explain this increase is that the parasite burden is low in those countries. IgE evolved as a host defense against parasites. In regions where the parasite burden is high, IgE is used for host defense against those organisms. But in developed regions where the parasite burden is low, IgE is available to cause allergic diseases. This is called the “hygiene” hypothesis, which states that people who live in countries with a high parasite burden have fewer allergic diseases, whereas those who live in countries with a low parasite burden have more allergic diseases.
The symptoms of these atopic disorders are induced by exposure to the specific allergens. These antigens are typically found in the environment (e.g., pollens released by plants and dust mite feces often found in bedding and carpet) or in foods (e.g., shellfish, eggs, and nuts). Exposure of nonatopic individuals to these substances does not elicit an allergic reaction. Many sufferers give immediate-type reactions to skin tests (injection, patch, or scratch) containing the offending antigen.
Atopic hypersensitivity is transferable by serum (i.e., it is antibody-mediated), not by lymphoid cells. In the past, this observation was used for diagnosis in the passive cutaneous anaphylaxis (Prausnitz-Küstner) reaction, which consists of taking serum from the patient and injecting it into the skin of a normal person. Some hours later, the test antigen, injected into the “sensitized” site, will yield an immediate wheal-and-flare reaction. This test is now impractical because of the danger of transmitting certain viral infections. Radioallergosorbent tests (RAST) permit the identification of specific IgE against potentially offending allergens if suitable specific antigens for in vitro tests are available.
There is evidence that initiation of the atopic response occurs when proteases in allergens, such as fungal allergens, pollens, and dust mite feces, cleave fibrinogen. The resulting cleavage products then activate Toll-like receptors (TLR-4) on the surface of macrophages and airway-lining cells to activate the atopic response.
Several genes associated with atopy have been identified. Mutations in the gene encoding the alpha chain of the IL-4 receptor strongly predispose to atopy. These mutations enhance the effectiveness of IL-4, resulting in an increased amount of IgE synthesis by B cells. Other genes identified include the gene for IL-4 itself, the gene for the receptor for the epsilon heavy chain, and several class II major histocompatibility complex (MHC) genes.
Drug Hypersensitivity
Drugs, particularly antimicrobial agents such as penicillin, are now among the most common causes of hypersensitivity reactions. Usually it is not the intact drug that induces antibody formation. Rather, a metabolic product of the drug, which acts as a hapten and binds to a body protein, does so. The resulting antibody can react with the hapten or the intact drug to give rise to type I hypersensitivity.2
When reexposed to the drug, the person may exhibit a drug rash, fever, or local or systemic anaphylaxis of variable severity. Reactions to very small amounts of the drug can occur (e.g., in a skin test with the hapten). A clinically useful example is the skin test using penicilloyl polylysine to reveal an allergy to penicillin.
Desensitization
Major manifestations of anaphylaxis occur when large amounts of mediators are suddenly released as a result of a massive dose of antigen abruptly combining with IgE on many mast cells. This is systemic anaphylaxis, which is potentially fatal. Desensitization can prevent systemic anaphylaxis.
Acute desensitization involves the administration of very small amounts of antigen at 15-minute intervals. Antigen–IgE complexes form on a small scale, and not enough mediator is released to produce a major reaction. This permits the administration of a drug or foreign protein to a hypersensitive person, but the hypersensitive state returns because IgE continues to be made.
Chronic desensitization involves the long-term weekly administration of the antigen to which the person is hypersensitive. This stimulates the production of IgA-and IgG-blocking antibodies, which can prevent subsequent antigen from reaching IgE on mast cells, thus preventing a reaction. It also induces regulatory T cells to produce IL-10, which reduces the synthesis of IgE.
Treatment & Prevention
Treatment of anaphylactic reactions includes drugs to counteract the action of mediators, maintenance of an airway, and support of respiratory and cardiac function. Epinephrine, antihistamines, corticosteroids, or cromolyn sodium, either singly or in combination, should be given. Cromolyn sodium prevents release of mediators (e.g., histamine) from mast cell granules. Prevention relies on identification of the allergen by a skin test and avoidance of that allergen.
There are several approaches to the treatment of asthma. Inhaled β-adrenergic bronchodilators, such as albuterol, are commonly used. Corticosteroids, such as prednisone, are also effective. Aminophylline, a bronchodilator, is effective but not commonly used. A monoclonal anti-IgE antibody (omalizumab, Xolair) is indicated for patients with severe asthma whose symptoms are not controlled by corticosteroids. For the prevention of asthma, leukotriene receptor inhibitors, such as montelukast (Singulair), and cromolyn sodium are effective.
The treatment of allergic rhinitis typically involves antihistamines along with nasal decongestants. For allergic conjunctivitis, eye drops containing antihistamines or vasoconstrictors are effective. Avoidance of the inciting allergens, such as pollens, is helpful in prophylaxis. Desensitization can also be helpful.
TYPE II: CYTOTOXIC HYPERSENSITIVITY
Cytotoxic hypersensitivity occurs when antibody directed at antigens of the cell membrane activates complement (Figure 65–2). This generates a membrane attack complex (see Chapter 63), which damages the cell membrane. The antibody (IgG or IgM) attaches to the antigen via its Fab region and acts as a bridge to complement via its Fc region. As a result, there is complement-mediated lysis as in hemolytic anemias, ABO transfusion reactions, or Rh hemolytic disease. In addition to causing lysis, complement activation attracts phagocytes to the site, with consequent release of enzymes that damage cell membranes.
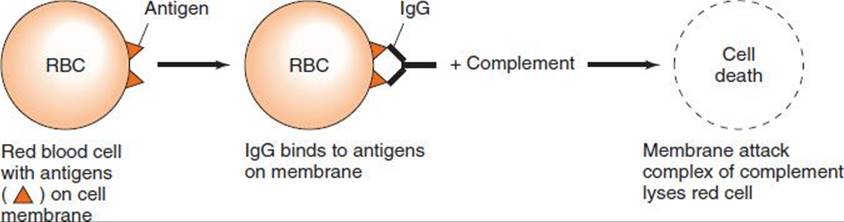
FIGURE 65–2 Cytotoxic hypersensitivity. RBC, red blood cell.
Drugs (e.g., penicillins, phenacetin, quinidine) can attach to surface proteins on red blood cells and initiate antibody formation. Such autoimmune antibodies (IgG) then interact with the red blood cell surface and result in hemolysis. The direct antiglobulin (Coombs) test is typically positive (see Chapter 64). Other drugs (e.g., quinine) can attach to platelets and induce autoantibodies that lyse the platelets, producing thrombocytopenia and, as a consequence, a bleeding tendency. Others (e.g., hydralazine) may modify host tissue and induce the production of autoantibodies directed at cell DNA. As a result, disease manifestations resembling those of systemic lupus erythematosus occur. Certain infections (e.g., Mycoplasma pneumoniae infection) can induce antibodies that cross-react with red cell antigens, resulting in hemolytic anemia. In rheumatic fever, antibodies against the group A streptococci cross-react with cardiac tissue. In Goodpasture’s syndrome, antibody to basement membranes of the kidneys and lungs bind to those membranes and activate complement. Severe damage to the membranes is caused by proteases released from leukocytes attracted to the site by complement component C5a (see page 529).
TYPE III: IMMUNE COMPLEX HYPERSENSITIVITY
Immune complex hypersensitivity occurs when antigen–antibody complexes induce an inflammatory response in tissues (Figure 65–3). Normally, immune complexes are promptly removed by the reticuloendothelial system, but occasionally they persist and are deposited in tissues, resulting in several disorders. In persistent microbial or viral infections, immune complexes may be deposited in organs (e.g., the kidneys), resulting in damage. In autoimmune disorders, “self” antigens may elicit antibodies that bind to organ antigens or deposit in organs as complexes, especially in joints (arthritis), kidneys (nephritis), or blood vessels (vasculitis).
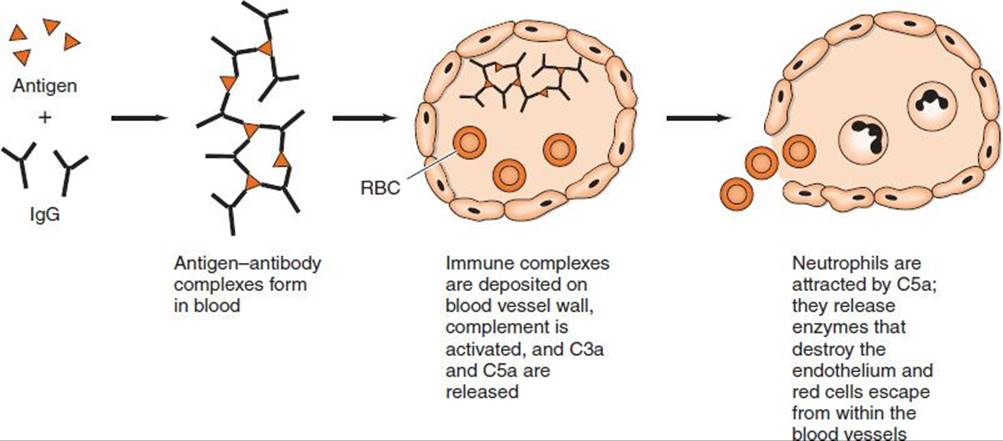
FIGURE 65–3 Immune complex hypersensitivity. RBC, red blood cell.
Wherever immune complexes are deposited, they activate the complement system. Polymorphonuclear cells are attracted to the site, and inflammation and tissue injury occur. Two typical type III hypersensitivity reactions are the Arthus reaction and serum sickness.
Arthus Reaction
Arthus reaction is the name given to the inflammation caused by the deposition of immune complexes at a localized site. It is named for Dr. Arthus, who first described the inflammatory response that occurs under the following conditions. If animals are given an antigen repeatedly until they have high levels of IgG antibody3 and that antigen is then injected subcutaneously or intradermally, intense edema and hemorrhage develop, reaching a peak in 3 to 6 hours.
Antigen, antibody, and complement are deposited in vessel walls; polymorphonuclear cell infiltration and intravascular clumping of platelets then occur. These reactions can lead to vascular occlusion and necrosis.
A clinical manifestation of the Arthus reaction is hypersensitivity pneumonitis (allergic alveolitis) associated with the inhalation of thermophilic actinomycetes (“farmer’s lung”) growing in plant material such as hay. There are many other occupation-related examples of hypersensitivity pneumonitis, such as “cheese-worker’s lung,” “woodworker’s lung,” and “wheat-miller’s lung.” Most of these are caused by the inhalation of some microorganism, either bacterium or fungus, growing on the starting material. An Arthus reaction can also occur at the site of tetanus immunizations if they are given at the same site with too short an interval between immunizations. (The minimum interval is usually 5 years.)
Serum Sickness
In contrast to the Arthus reaction, which is localized inflammation, serum sickness is a systemic inflammatory response to the presence of immune complexes deposited in many areas of the body. After the injection of foreign serum (i.e., serum from another animal such as a horse [or, more commonly these days, exposure to certain drugs]), the antigen is excreted slowly. During this time, antibody production starts. The simultaneous presence of antigen and antibody leads to the formation of immune complexes, which may circulate or be deposited at various sites.
Typical serum sickness results in fever, urticaria, arthralgia, lymphadenopathy, splenomegaly, and eosinophilia a few days to 2 weeks after injection of the foreign serum or drug. Although it takes several days for symptoms to appear, serum sickness is classified as an immediate reaction because symptoms occur promptly after immune complexes form. Symptoms improve as the immune system removes the antigen and subside when the antigen is eliminated. Nowadays, serum sickness is caused more commonly by drugs (e.g., penicillin) than by foreign serum because foreign serum is used so infrequently. A maculopapular drug-induced rash to penicillins, such as ampicillin, is quite common. Use of antithymocyte globulin (thymoglobulin), which is made in horses, to provide immunosuppression in transplant patients may cause serum sickness. Note also that diphtheria antitoxin made in horses is known to cause serum sickness.
Immune Complex Diseases
Many clinical disorders associated with immune complexes have been described, although the antigen that initiates the disease is often in doubt. Several representative examples are described next.
Glomerulonephritis
Acute poststreptococcal glomerulonephritis is a well-accepted immune complex disease. Its onset follows several weeks after a group A β-hemolytic streptococcal infection, particularly of the skin, and often with nephritogenic serotypes of Streptococcus pyogenes. Typically, the complement level is low, suggesting an antigen–antibody reaction. Lumpy deposits of immunoglobulin and C3 are seen along glomerular basement membranes by immunofluorescence, suggesting the presence of antigen–antibody complexes. It is assumed that streptococcal antigen–antibody complexes, after being deposited on glomeruli, fix complement and attract neutrophils, which start the inflammatory process.
Similar lesions with “lumpy” deposits containing immunoglobulin and C3 occur in infective endocarditis, serum sickness, and certain viral infections (e.g., hepatitis B and dengue hemorrhagic fever). Lesions containing immune complexes also occur in autoimmune diseases (e.g., the nephritis of systemic lupus erythematosus, in which the “lumpy” deposits contain DNA as the antigen) (see later and page 556).
IgA nephropathy is one of the most common forms of immune complex glomerulonephritis worldwide. This disease is characterized by deposits of IgA on the glomeruli. The cause is unknown; no infectious agent has been associated with this disease. The course of the disease varies widely. Some patients are asymptomatic, some have mild symptoms, and others progress rapidly to kidney failure. Diagnosis is made by doing renal biopsy and demonstrating IgA deposits by immunohistologic testing.
Rheumatoid Arthritis
Rheumatoid arthritis is a chronic inflammatory autoimmune disease of the joints seen commonly in young women. It is a systemic disease involving not only the joints but other organs as well, most often the lung and pericardium. Serum and synovial fluid of patients contain “rheumatoid factor” (i.e., IgM and IgG antibodies that bind to the Fc fragment of normal human IgG). Deposits of immune complexes (containing the normal IgG and rheumatoid factor) on synovial membranes and in blood vessels activate complement and attract polymorphonuclear cells, causing inflammation. Patients have high titers of rheumatoid factor and low titers of complement in serum especially during periods when their disease is most active (see page 556).
Systemic Lupus Erythematosus
Systemic lupus erythematosus is a chronic inflammatory autoimmune disease that affects several organs, especially the skin of the face, the joints, and the kidneys. Antibodies are formed against DNA and other components of the nucleus of cells. These antibodies form immune complexes that activate complement. Complement activation produces C5a, which attracts neutrophils that release enzymes, thereby damaging tissue (see pages 529 and 556).
TYPE IV: DELAYED (CELL-MEDIATED) HYPERSENSITIVITY
Delayed hypersensitivity is a function of T lymphocytes, not antibody (Figure 65–4). It can be transferred by immunologically committed (sensitized) T cells, not by serum. The response is “delayed” (i.e., it starts hours [or days] after contact with the antigen and often lasts for days).
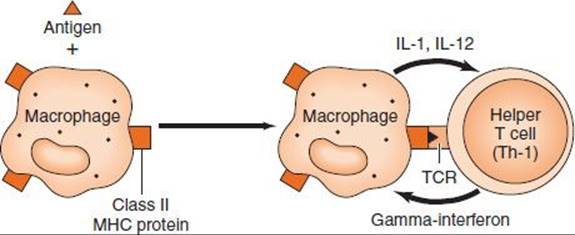
FIGURE 65–4 Delayed (cell-mediated) hypersensitivity. The macrophage ingests the antigen, processes it, and presents an epitope on its surface in association with class II major histocompatibility complex (MHC) protein. The helper T (Th-1) cell is activated and produces gamma interferon, which activates macrophages. These two types of cells mediate delayed hypersensitivity. TCR, T-cell receptor.
In certain contact hypersensitivities, such as poison oak, the pruritic, vesicular skin rash is caused by CD8-positive cytotoxic T cells that attack skin cells that display the plant oil as a foreign antigen. In the tuberculin skin test, the indurated skin rash is caused by CD4-positive helper T cells and macrophages that are attracted to the injection site. Table 65–4 describes some of the important clinical aspects of delayed hypersensitivities.
TABLE 65–4 Important Clinical Aspects of Delayed Hypersensitivities
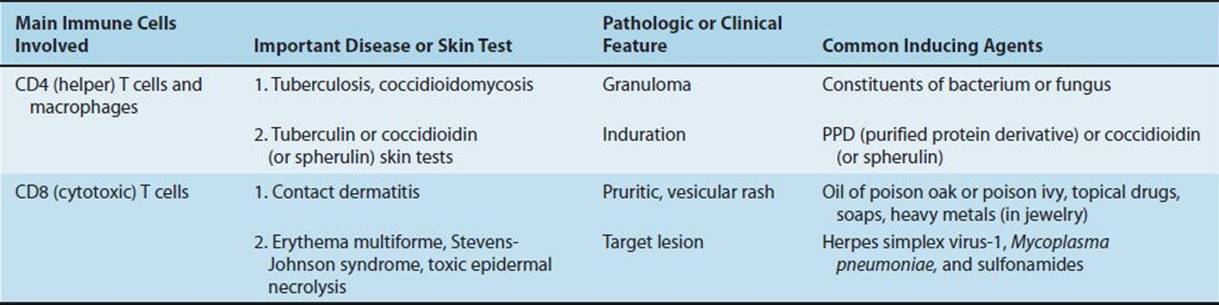
Clinically Important Delayed Hypersensitivity Reactions
Contact Hypersensitivity
This manifestation of cell-mediated hypersensitivity occurs after sensitization with simple chemicals (e.g., nickel, formaldehyde), plant materials (e.g., poison ivy, poison oak), topically applied drugs (e.g., sulfonamides, neomycin), some cosmetics, soaps, and other substances. Neomycin in topical antibacterial ointment is a very common cause.
In all cases, the small molecules acting as haptens enter the skin, attach to body proteins, and become complete antigens. It is thought that these normal skin proteins to which the immune system is tolerant now can act as a carrier protein, because the hapten alters the protein enough that the immune system recognizes it as foreign. Cell-mediated hypersensitivity is induced, particularly in the skin. Upon a later skin contact with the offending agent, the sensitized person develops contact dermatitis characterized by erythema, itching, vesicles, eczema, or necrosis of skin within 12 to 48 hours caused by the attack of cytotoxic T cells. Patch testing on a small area of skin can sometimes identify the offending antigen. Subsequent avoidance of the material will prevent recurrences.
Tuberculin-Type Hypersensitivity
Delayed hypersensitivity to antigens of microorganisms occurs in many infectious diseases and has been used as an aid in diagnosis. It is typified by the tuberculin reaction. When a patient previously exposed to Mycobacterium tuberculosis is injected with a small amount of tuberculin (purified protein derivative [PPD]) intradermally, there is little reaction in the first few hours. Gradually, however, induration and redness develop and reach a peak in 48 to 72 hours. A positive skin test indicates that the person has been infected with the agent, but it does not confirm the presence of current disease. However, if the skin test converts from negative to positive, it suggests that the patient has been recently infected. Infected persons do not always have a positive skin test, because overwhelming infection, disorders that suppress cell-mediated immunity (e.g., uremia, measles, sarcoidosis, lymphoma, and acquired immunodeficiency syndrome [AIDS]), or the administration of immunosuppressive drugs (e.g., corticosteroids, antineoplastics) may cause anergy.
A positive skin test response assists in diagnosis and provides support for chemoprophylaxis or chemotherapy. In leprosy, a positive lepromin test indicates the presence of tuberculoid leprosy with competent cell-mediated immunity, whereas a negative lepromin test suggests the presence of lepromatous leprosy with impaired cell-mediated immunity. In systemic mycotic infections (e.g., coccidioidomycosis and histoplasmosis), a positive skin test with the specific antigen indicates exposure to the organism. Cell-mediated hypersensitivity develops in many viral infections; however, serologic tests are more specific than skin tests both for diagnosis and for assessment of immunity. In protozoan and helminthic infections, skin tests may be positive, but they are generally not as useful as specific serologic tests.
Erythema Multiforme, Stevens-Johnson Syndrome, and Toxic Epidermal Necrolysis
Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis are related skin diseases caused primarily by cytotoxic T-cell attack on skin cells (keratinocytes). The most common triggers are herpes simplex virus-1, M. pneumoniae, and a variety of drugs, including sulfonamides and penicillins. Several human leukocyte antigen (HLA) alleles predispose to these diseases, especially HLA-DQ3 and HLA-B12.
The clinical manifestations of these diseases are characterized by a continuum of symptoms that differ in severity and anatomic location. Erythema multiforme minor is characterized by relatively few, localized target lesions on the skin, often involving the extremities (Figure 65–5), with minimal involvement of mucous membranes. They begin to heal in 7 days but may recur. In contrast, erythema multiforme major has more extensive lesions on the skin and involves the mucous membranes, often of the mouth and conjunctivae.
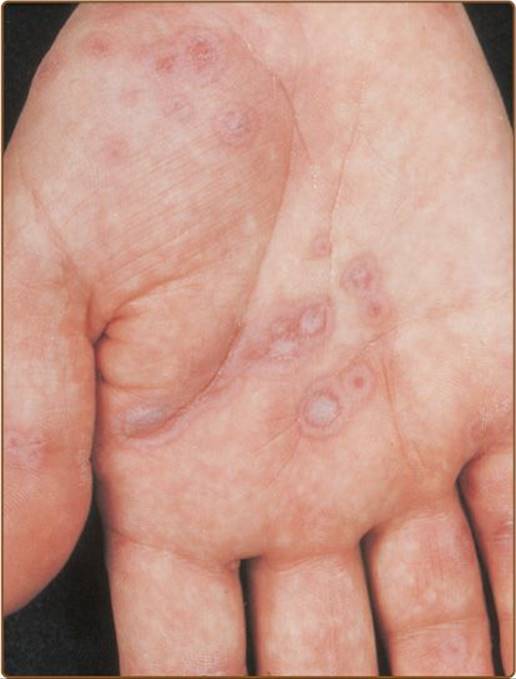
FIGURE 65–5 Erythema multiforme. Target lesions on palm. (Reproduced with permission from Goldsmith LA, Katz SI et al (eds). Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York: McGraw-Hill, 2012. Copyright © 2012 by The McGraw-Hill Companies, Inc.)
Stevens-Johnson syndrome (SJS) has more extensive blistering lesions, often on the face and trunk with significant lesions on the mucous membranes. In SJS, less than 10% of the body surface is involved; in toxic epidermal necrolysis (TEN), more than 10% of the body surface is involved. TEN is a life-threatening disease, and treatment in a burn unit is recommended.
SELF-ASSESSMENT QUESTIONS
1. Your patient has episodes of eye tearing, “blood-shot” eyes, and runny nose, which you think may be due to an allergy to some plant pollen. You refer the patient to an allergist, who performs skin tests with various allergens. A wheal-and-flare reaction is seen on the patient’s back at the site where several pollens were injected. What is the most likely sequence of events that produced the wheal-and-flare reaction?
(A) Allergen binds to IgE on the surface of B cells and IL-4 is released.
(B) Allergen binds to IgE on the surface of mast cells and histamine is released.
(C) Allergen binds to IgE in the plasma, which activates complement to produce C3b.
(D) Allergen binds to IgE in the plasma, and the allergen-IgE complex binds to the surface of macrophages and IL-1 is released.
2. One important test to determine whether your patient has been exposed to Mycobacterium tuberculosis, the organism that causes tuberculosis, is to do a PPD skin test. In this test, PPD extracted from the organism is injected intradermally. Of the following, which one is most likely to occur at the site of a positive PPD?
(A) Cytotoxic T cells kill target cells at the site.
(B) Macrophages and CD4-positive T cells infiltrate the site.
(C) Histamine and leukotrienes are liberated from mast cells at the site.
(D) Immune complexes consisting of PPD and IgG are deposited at the site.
3. Your patient is a 77-year-old man with enterococcal endocarditis who was treated with penicillin G and gentamicin. Five days later, fever and a diffuse maculopapular rash developed. There is no urticaria, hypotension, or respiratory compromise. Urinalysis revealed proteinuria and granular casts. You suspect he may have serum sickness. Which one of the following immunopathogenic mechanisms is most likely to be the cause?
(A) One of the drugs formed immune complexes with IgG.
(B) One of the drugs activated CD4-positive T cells and macrophages.
(C) One of the drugs activated the alternative pathway of complement.
(D) One of the drugs cross-linked IgE on the mast cells and caused the release of histamine.
4. Of the following diseases, which one is most likely to be caused by a delayed hypersensitivity reaction?
(A) Autoimmune hemolytic anemia
(B) Contact dermatitis, such as poison oak
(C) Hemolytic disease of the newborn
(D) Poststreptococcal glomerulonephritis
(E) Systemic lupus erythematosus
5. Atopic individuals (i.e., those with a hereditary predisposition to immediate hypersensitivity reactions) produce an increased amount of IgE. Of the following, which one is the most likely explanation for the increased production of IgE?
(A) Large amounts of IL-1 are produced by dendritic cells.
(B) Large amounts of IL-2 are produced by macrophages.
(C) Large amounts of IL-4 are produced by Th-2 cells.
(D) Large amounts of gamma interferon are produced by Th-1 cells.
(E) Large amounts of C3a are produced by the alternative pathway of complement.
6. Of the following four types of hypersensitivity reactions, which one causes the hemolysis that occurs in hemolytic disease of the newborn (erythroblastosis fetalis)?
(A) Type I–immediate hypersensitivity
(B) Type II–cytotoxic hypersensitivity
(C) Type III–immune complex hypersensitivity
(D) Type IV–delayed hypersensitivity
ANSWERS
1. (B)
2. (B)
3. (A)
4. (B)
5. (C)
6. (B)
PRACTICE QUESTIONS: USMLE & COURSE EXAMINATIONS
Questions on the topics discussed in this chapter can be found in the Immunology section of PART XIII: USMLE (National Board) Practice Questions starting on page 713. Also see PART XIV: USMLE (National Board) Practice Examination starting on page 731.
1An increase in cyclic guanosine monophosphate (GMP) within these cells increases mediator release, whereas an increase in cyclic adenosine monophosphate (AMP) decreases the release. Therefore, drugs that increase intracellular cyclic AMP, such as epinephrine, are used to treat type I reactions. Epinephrine also has sympathomimetic activity, which is useful in treating type I reactions.
2Some drugs are involved in cytotoxic hypersensitivity reactions (type II) and in serum sickness (type III).
3Much more antibody is typically needed to elicit an Arthus reaction than an anaphylactic reaction.