Robert K. Semple
Learning objectives
After reading this chapter you should understand:
![]() The principles governing endocrine signaling and its regulation.
The principles governing endocrine signaling and its regulation.
![]() The anatomy, organization and regulatory role of the hypothalamus and pituitary.
The anatomy, organization and regulatory role of the hypothalamus and pituitary.
![]() The processes controlling biosynthesis, transport and action of thyroid hormones.
The processes controlling biosynthesis, transport and action of thyroid hormones.
![]() The mechanisms regulating synthesis and action of glucocorticoids.
The mechanisms regulating synthesis and action of glucocorticoids.
![]() The regulation of the synthesis and activity of sex steroid hormones and their role in human reproduction.
The regulation of the synthesis and activity of sex steroid hormones and their role in human reproduction.
![]() The direct and indirect actions of growth hormone, including the role of IGF-I.
The direct and indirect actions of growth hormone, including the role of IGF-I.
![]() The role of prolactin in reproduction.
The role of prolactin in reproduction.
![]() The consequences of deficiencies and excesses of hormones regulated by the hypothalamo-pituitary axis.
The consequences of deficiencies and excesses of hormones regulated by the hypothalamo-pituitary axis.
Introduction
As a first approximation, the human body contains 10 trillion – or 10 million million – cells, which may lie more than 2 meters apart. Coordination and regulation of the growth and multiple functions of these cells in order to meet the demands of existence in constantly fluctuating environmental conditions require precise and sophisticated integrative systems. The nervous system is a key part of the solution, being particularly well suited to mediating reflexes and motor actions needed, for example, in ‘fight or flight’ situations, which may require responses in fractions of a second. The endocrine system, however, is the principal means of regulation of a wide range of less acute functions including growth, development, reproduction, many aspects of homeostasis, and the response to more chronic external stimuli and stress. Such endocrine responses occur over several seconds at their fastest, to days or weeks at their slowest. Failures in these highly elaborate, complex and often interconnected endocrine control systems are common, and lead to many highly prevalent diseases.
Hormones
Hormones are chemical substances that are produced by particular glands or groups of cells, and that elicit specific responses from distant cells or organs
The concept that organs and tissues may produce ‘internal secretions’ that influence the behavior of distant parts of the body probably first emerged in France in the 18th century, and by the beginning of the 20th century, adrenaline and vasopressin (antidiuretic hormone, ADH) had been isolated. However, it was only in 1905, shortly after isolating secretin, that Ernest Starling in London first coined the word ‘hormone’ to describe such internal messengers. Hormones are now understood to be chemical substances that are produced by particular glands or groups of cells, and that elicit specific responses from distant cells or organs. Classically, such hormones are blood-borne and are described as endocrine hormones. However, subsequently it has become clear that some hormones act locally on cells around their cell of origin – so-called paracrine hormones – or even on the same cell that has produced them – so-called autocrine hormones.
Types of hormones
Hormones have diverse chemical structures
Many different types of molecules function as hormones (Table 39.1). Among the most chemically simple of hormones are modified amino acids, such as epinephrine (adrenaline), and many such simple amines also function as neurotransmitters (Chapter 41.1). At the other end of the spectrum, some hormones are polypeptides varying in size from tripeptides (e.g. thyrotropin-releasing hormones) to complex glycoproteins (e.g. luteinizing hormone, LH). The smaller peptide hormones are synthesized as large polypeptide prohormones, that are cleaved by proteolytic enzymes to release active hormone from the endocrine gland. Sometimes one prohormone can be cleaved in different ways to produce many different hormones, according to the tissue and cellular context. Other hormones are derived by modification of simple lipids such as cholesterol or fatty acids.
Table 39.1
Chemical derivation of hormones

AVP/ADH, arginine vasopressin or antidiuretic hormone; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
Many glands and tissues produce hormones
Hormones are classically known to be secreted from ductless glands such as the thyroid, adrenals, and pituitary which have been known to anatomists for centuries. However, many hormones are so potent that they need only circulate at tiny (e.g. picomolar) plasma concentrations to exert meaningful biological effects, and may derive from small clusters of cells, or from one particular cell type scattered within a larger organ. Furthermore, it is now becoming clear that many tissues which were not traditionally thought to be active in endocrine terms actually secrete a wide variety of different hormones with diverse effects on other tissues. Indeed, the availability of the human genome sequence, allied to sophisticated bio-informatics and techniques such as microarray-based transcriptional profiling (Chapter 36), has led to the discovery of many new suspected hormones and hormone receptors, and understanding of the endocrine mechanisms regulating human physiology is likely to increase rapidly. Nevertheless, in this chapter the emphasis is on the very well-established hormonal systems which are the focus of current clinical endocrinology.
![]() Advanced concept box Nonclassic endocrine organs
Advanced concept box Nonclassic endocrine organs
Many tissues apart from classic ductless endocrine glands are now known to be active endocrinologically, and the produced signals mediate important metabolic cross-talk between different tissues. Examples of this include production of leptin from white adipose tissue, which signals body energy stores to the brain (Chapter 22), production of fibroblast growth factor 23 (FGF23) from bone, (which modulates renal phosphate handling and is ectopically produced by some tumors, resulting in a clinically important renal phosphate leak and osteomalacia), and production of satiety factors such as ghrelin from the small intestine. The placenta is also a highly active endocrine tissue: as well as producing βHCG, progesterone, placental growth hormone and human placental lactogen, which have well-understood roles in pregnancy; it elaborates many other hormones at high levels whose role remains to be determined.
Principles of hormone action
In order to function effectively to relay signals from one part of the body to others, endocrine systems must exhibit certain general properties (Fig. 39.1).

FIG. 39.1 Basic endocrine processes.
The feedback regulation of hormone action is a classic example of self-regulation. Negative feedback regulation is common. Positive feedback (the feed-forward loop) is rare. Note how the feedback loops operate at different levels of the endocrine system.
Coupling of hormone release to relevant stimuli
Some hormones function to transmit to the body important environmental cues, while others are more concerned with calibrating metabolic processes involved in maintaining homeostasis
In either situation, it is critical that there is effective translation of the magnitude of the stimulus into the amount of hormone released. How this is achieved varies depending on the stimulus. Simple homeostatic sensing mechanisms include direct coupling of parathyroid hormone release to calcium levels via a cell surface receptor which binds calcium (Chapter 26), and direct coupling of insulin secretion to plasma glucose levels via sensing of blood glucose concentration by the pancreatic β cell (Chapter 21). Many of the endocrine systems described in this chapter, in contrast, are controlled by modified neurons which secrete hormones into the bloodstream. The level of secretion of such neurons is often determined by complex synaptic inputs from many different parts of the brain, as well as by the sensing of circulating metabolites and hormones. These secretory neurons thus function as important sites of integration of a variety of environmental, psychologic and physiologic cues.
Feedback regulation: a major feature of endocrine systems
A major feature of most endocrine systems is negative feedback (Fig. 39.1). This means that a response induced by action of a hormone feeds back to inhibit the level of hormone production. The effect of this is to damp down fluctuations in the process controlled by the hormone, thereby enhancing stability of that process and hence homeostasis. However, were negative feedback to be the only process at play, then the output of the system would remain constant. In fact, many endocrine systems, notably those controlled by the hypothalamus, display distinct rhythmicity. This is determined by the net product of the neural inputs to the relevant secretory neurons, and in this situation negative feedback serves instead to smooth out the hormonal profile and to prevent short-term instability. Furthermore, modulation of the susceptibility of the controlling neuron to feedback inhibition represents one potential mechanism of modulating the level of the output of the endocrine system.
Positive feedback refers to stimulation of hormone release by the response it provokes. This feed-forward loop is inherently unstable, and leads to a rapid, exponential increase in the level of signal. This is much rarer than negative feedback in physiology, but plays an important role in some processes which require precise timing, such as the luteinizing hormone surge which drives ovulation.
Transduction of signal at target tissues: hormone receptors
Hormones act by binding to specific receptors, either on the cell surface or within the target cell (Chapter 40)
Classically, there is a very high degree of specificity in this binding, and it is this hormone–receptor interaction that triggers and coordinates a wide range of biological effects. Hormone receptors may be divided into different families according to their structure, and each family broadly shares signal transduction mechanisms.
Many amine-like hormones and peptides act via receptors that are intrinsic plasma membrane proteins
Many amine-like hormones and peptides which cannot cross lipid bilayers act via G-protein coupled receptors, while some larger peptides act via tyrosine kinase receptors (e.g. insulin and IGF-I) or cytokine-like receptors (e.g. leptin and growth hormone). All of these receptor types are intrinsic plasma membrane proteins, and rely on cascades of sequential phosphorylation events to alter enzyme activity and gene expression.
Lipophilic hormones enter cells before binding their receptors
In contrast lipophilic hormones including steroid hormones, some hormones derived from fatty acids, and tri-iodothyronine, which has physicochemical properties in common with steroids (Chapter 17), enter cells before binding to their receptors. These so-called nuclear hormone receptors are effectively ligand-activated transcription factors, and provide a very direct link between the exposure of a cell to hormonal ligand and alteration of patterns of gene expression (Chapter 35).
Many putative receptors for unknown hormones have been identified by virtue of sequence homology among members of each of the major hormone receptor classes. It is likely that identification of the ligand for these ‘orphan’ receptors will significantly increase the range of endocrine hormones. Such putative receptor-encoding genes are also a major focus of pharmaceutical research, due to the relative ease with which receptors may be targeted by novel drugs.
Turning off the hormonal signal
Levels of hormones are useful physiologic signals only if there is also an effective way of turning the signals off
Hormone inactivation usually occurs by further metabolism (e.g. proteolysis of peptides, or hydroxylation, conjugation and excretion of many steroids). Such degradation may occur in plasma, in organs such as the liver, or in target tissues after receptor-mediated internalization of the hormone. The rate of clearance of different hormones varies enormously, from a few minutes (insulin), through hours (steroids) to days (thyroxine). Measurement of urinary concentrations of hormones or their metabolites can be used in some diagnostic settings, including the diagnosis of pregnancy, and in pathologies such as secretory adrenal tumors.
![]() Advanced concept box Preservation of epigenetic ‘marks’ (imprinting) in endocrinology
Advanced concept box Preservation of epigenetic ‘marks’ (imprinting) in endocrinology
Gene expression may be influenced not only by the DNA sequence of genes and their promoter regions, but also by covalent modifications of DNA and/or histones.Methylation of cytosine residues occurs particularly in promoter regions and acts to silence genes, for example during tissue differentiation. During the earliest stages of development most of these epigenetic ‘marks’ are wiped clean to generate totipotent embryonic stem cells. However, they are preserved at some genes, and this pattern of covalent modifications often shows which parent the gene was inherited from. Because epigenetic marks are preserved as cells divide, major ‘parent of origin’ effects can be seen subsequently on gene expression in some or all tissues.
Several endocrine systems show significant imprinting. Best known is the IGF2-IGF2 receptor system, which acts to influence placental and fetal growth, in common with nearly all known imprinted genes. Another important endocrine signaling gene which is imprinted is the GNAS gene, encoding a stimulatory G-protein subunit coupled to many classic endocrine peptide hormone receptors. Various different mutations have been described, and depending on which parent they are inherited from, they may produce skeletal dysplasia and resistance to various hormones, including most notably parathyroid hormone. Indeed, so-called ‘pseudohypoparathyroidism’ was the first reported example of end-organ resistance to a hormone. For review of the fascinating and complex biology of GNAS, readers are referred to Weinstein LS et al. Minireview: GNAS: normal and abnormal functions. Endocrinology 2004;145:5459–5464.
Carrier proteins and ‘free’ hormones
Many small or hydrophobic hormones are transported in plasma bound to carrier proteins
Within the circulation, many small or hydrophobic hormones are transported bound to carrier proteins. For example, thyroxine and cortisol are transported on plasma proteins such as thyroid-binding globulin (TBG) and cortisol-binding globulins (CBG). This has several consequences of relevance to both physiology and clinical measurement. The transport proteins extend the biological half-life and increase the plasma concentration of the smaller hormones, which would otherwise be eliminated rapidly in the liver or kidney. This also means that there may be large differences between the total plasma levels of hormone and the unbound or ‘free’ hormone in solution, which is generally the biologically active form. Thus clinical interpretation of plasma hormone levels is simplest if assays are developed to detect free hormone only, otherwise assumptions must be made about levels of binding proteins, which in turn may be significantly influenced by the nutritional and hormonal milieu. A common example of the problem posed by variable levels of carrier proteins comes in assessing cortisol levels in women taking estrogen-containing contraceptive pills, which stimulate binding protein levels and thus make the usual laboratory normal ranges for cortisol inapplicable. It is not only small lipophilic hormones which have plasma-binding proteins, however; both growth hormone and insulin-like growth factor 1 have plasma binding proteins which influence bio-availability and tissue activity of the hormones in a variety of different ways (Table 39.2).
Table 39.2
Major hormone binding proteins

Local metabolism of hormones at target tissues
Another feature of some endocrine systems, particularly those involving steroid hormones, is local metabolism of the hormones or prohormones near their cognate receptor
This may involve conversion of an active circulating hormone to a more potent form (for example, testosterone to dihydrotestosterone by 5α-reductase in androgen-dependent hair follicles and prostate gland), or creation of an active hormone from what is effectively a circulating prohormone (e.g. thyroxine to tri-iodothyronine by de-iodinase, or synthesis of estradiol from adrenal androgens in adipose tissue). A similar mechanism is also employed to prevent mineralocorticoid receptors in the kidney being exposed to high concentrations of cortisol, with cortisol being inactivated by 11β-hydroxysteroid dehydrogenase type II, which is expressed highly near the mineralocorticoid receptors. Many other examples of such intracellular interconversion of steroid hormones have also emerged, and study of this phenomenon has sometimes been dubbed ‘intracrinology’ (Table 39.3).
Table 39.3
Examples of hormone metabolism in target tissues

Several hormones may control one process, or one hormone may control several processes
While it may be convenient to think of the endocrine system as being compartmentalized, so that one hormone has control over one process, this is rarely the case
For example, at least four different hormones are involved in the regulation of plasma glucose concentration (Chapter 21). Conversely, single hormones such as testosterone influence a range of metabolic processes.
All of these features of endocrine systems will be illustrated later in this chapter by some or all of the endocrine axes which pass through the pituitary, to be discussed.
Biochemical assessment of hormone action
Laboratory testing of endocrine systems aims first to determine whether the system is functioning abnormally, and second to localize the functional defect
Most commonly, this means that the level of the hormone that elicits the target tissue response is determined, usually by immunoassay (Fig. 39.2), together with measurement of one or more upstream trophic hormones, where they exist. The presence of negative feedback regulation means that the system will attempt to correct perturbations in levels of the effector hormone with compensatory changes in levels of trophic hormones. Thus assessment of at least two points in such an endocrine feedback loop is essential, and permits focusing of later diagnostic imaging on the relevant gland.

FIG. 39.2 The sandwich design for enzyme-linked immunosorbent assay (ELISA) for the measurement of hormone concentration.
(A) The amount of signal present on the microplate after washing off the serum and reagents is a measure of the concentration of a hormone. (B) Calibration curve is a series of measurements of samples with a known concentration of a hormone (calibrators). Subseqently the signal obtained from the unknown samples is compared to the standard curve, and the hormone concentration is read out.
Once the hormones of most interest to the clinical problem have been chosen, care needs to be used to ensure appropriate sampling. This should take into account whether the hormone is very unstable (for example, many small peptide hormones require blood samples to be taken into tubes containing a protease inhibitor which are kept on ice), and also whether it is produced with a circadian or other rhythm. For some hormones, such as thyroxine, which have very long half-life, timing of the sample is not critical, while for others, such as cortisol, standardized timing of the sample is essential.
Single measurements are meaningless in case of hormones with pulsatile secretion
A further factor to take into account in hormone testing is whether or not secretion of the hormone is pulsatile. Nearly all of the hypothalamic and pituitary hormones show some degree of pulsatility in secretion. In most cases this simply means that repeat testing in the case of mild abnormality is required, while for some hormones, such as growth hormone, which has both a short half-life and very striking secretory spikes, single measurements are often meaningless.
Because of these problems, endocrinologists commonly employ multiple testing to build up a profile of hormone levels at several points in the day. This can be an effective way of detecting subtle perturbations of the circadian rhythm, and can also lessen the impact of oscillatory secretion of the hormone.
Some hormone are measured after a relevant stimulus has been applied
A further approach is to use provocative testing. In other words, hormone levels are measured not just in the resting state, but also after a relevant stimulus has been applied, in an attempt to assess the maximal capacity of the endocrine gland or system being tested. Frequently the stimulus is a high dose of trophic hormone (e.g. ACTH, TRH, GnRH, etc.); however, it may instead be a metabolic challenge (e.g. with an oral glucose load or severe insulin-induced hypoglycemia to mimic stress). Examples of each of these tests are given in Table 39.4.
Table 39.4
Examples of commonly used provocative endocrine tests

H-P, hypothalomo-pituitary.
![]() Clinical test box Hormone immunoassay
Clinical test box Hormone immunoassay
Immunoassay is the most widely applied technique for measuring hormone levels. Antibodies are produced that bind to antigenic sites on the hormone. Ideally, such antibody should possess both high specificity and affinity for the hormone of interest, e.g. the β-subunit of glycoprotein hormones.
In the first immunoassays, the antibodies were obtained from the serum of animal species (e.g. rabbit or sheep) that had been immunized with human hormone preparations. Such antisera contained a range of different antibodies (polyclonal) capable of binding to different sites on the hormone antigen. However, antibody specificity and titer varied with the animal and duration of the immune response.
Today most immunoassays employ monoclonal antibodies, which are produced by fusion of spleen cells from an immunized mouse with a mouse myeloma cell line. Hybridoma cells may be cloned to produce a cell line that secretes a single antibody species for an indefinite period of time.
Many commercial producers have designed proprietary methods for quantifying the hormone–antibody interaction. One of the most widely used formats is the two-site (sandwich) immunometric or enzyme-linked immunosorbent assay (ELISA) assay outlined in Figure 39.2. This assay employs two antibodies binding to different epitopes on the hormone, one of which is labeled or modified to be capable of generating an optical signal. Spectrophotometric, fluorometric, luminescence and radiochemical reporter systems are in common use.
Not all immunoassays rely on labeled antibody: other approaches include nephelometry, in which appearance of light-scattering immune complexes is monitored spectrophotometrically.
Major types of endocrine pathology
Autoimmunity to the thyroid, adrenal and pancreatic islets accounts for more than 50% of all organ-specific autoimmune disease
Although each endocrine system may malfunction as a consequence of organ-specific damage or disease, for example relating to the anatomic site of the gland, there are also some general types of pathology to which endocrine cells are very susceptible. The first of these is autoimmune destruction with detectable endocrine-gland-specific antibodies. This is a potential cause of loss of function of nearly all endocrine glands and, less commonly, may also cause gland hyperfunction. In fact, autoimmunity to the thyroid, adrenal and pancreatic islets accounts for more than 50% of all organ-specific autoimmune disease. This appears in part due to genetic predisposition, accounted for by both MHC and non-MHC genes, and also by poorly defined environmental factors (Chapter 38).
The second major group of pathologies which present as endocrine disease is neoplasia, which may be either benign or malignant
Neoplastic cells arise as autonomous clones within endocrine glands due to somatic mutations in growth factor signaling pathways or commonly in G-protein coupled signaling pathways. They may produce disease due to either excessive and dysregulated production of biologically active hormones or damage to neighboring, normal endocrine cells, with attendant loss of hormone secretion. Very small and benign adenomas which would remain undetected in nonendocrine tissues may produce florid disease by virtue of hormone hypersecretion coupled to the potency of hormones in eliciting biological responses.
As well as the pathologies arising in endocrine cells or organs, the very nature of hormones, and their frequent availability in medical preparations, means that they may also cause clinical problems when used exogenously, either as a side effect of a desired therapeutic action (e.g. corticosteroids used in inflammatory disease causing weight gain and diabetes), or due to inappropriate self administration (e.g. thyroid hormone to induce weight loss).
The hypothalamo–pituitary regulatory system
Hormones of the posterior pituitary gland are distinct from those of the anterior pituitary
The pituitary gland is a pea-sized, oval organ encased in a bony cavity of the skull (the sella turcica) below the brain. It communicates with the hypothalamus via the pituitary stalk, which contains a complex array of axons and portal blood vessels (Fig. 39.3). The pituitary gland is divided into two lobes. The posterior pituitary (or neurohypophysis) is embryologically part of the brain, and consists largely of neurons which have cell bodies in the supraoptic and paraventricular nuclei of the hypothalamus. It is in these cell bodies that hormones are synthesized and packaged before transport on microtubules along axons to the pituitary, where they are released. The anterior lobe (adenohypophysis), accounting for approximately 80% of the gland, is embryologically derived from oral ectoderm, and has no direct anatomic continuity with the brain. Instead, it may be viewed as a target organ for endocrine hormones released from hypothalamic nuclei and transported from the median eminence by the portal circulation.
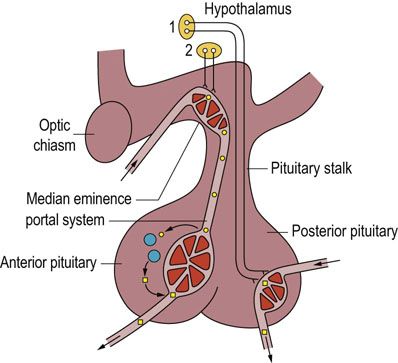
FIG. 39.3 The hypothalamo-anterior pituitary regulatory system.
Hormones of the posterior pituitary are synthesized and packaged in the supraoptic and paraventricular nuclei of the hypothalamus (1), transported along axons and stored in the posterior pituitary prior to release into the circulation. The anterior pituitary releasing- or release-inhibiting hormones are synthesized in various hypothalamic nuclei (2) and transported to the median eminence. From there they travel to the anterior pituitary via a portal venous system.
Both posterior and anterior pituitary are controlled largely by the hypothalamus
The hypothalamus is a highly connected center of the CNS, receiving synaptic inputs from vast numbers of different parts of the brain as well as sensing peripheral signals through areas of porosity in the blood–brain barrier. The hypothalamus thus functions as an integrative center which orchestrates a huge number of endocrine and neural processes, and entrains them to relevant external stimuli. The endocrine systems which involve the hypothalamus, pituitary and downstream organs are usually termed ‘axes’ and are most usefully viewed asfunctional units for the purposes of clinical diagnosis and management (Fig. 39.4).

FIG. 39.4 Hypothalamo-anterior pituitary-regulatory target organ axes.
The hypothalamo-anterior pituitary regulatory system comprises 5 parallel endocrine axes, regulating the biosynthesis and release of: (1) thyroid hormone; (2) glucocorticoids; (3) sex steroids; (4) growth hormone; and (5) prolactin. T4, thyroxine; T3, tri-iodothyronine; GHRH, growth hormone-releasing hormone; GnRH, gonadotropin-releasing hormone; IGF-I, insulin growth factor-I ACTH adrenocorticotropic hormone; FSH, follicle- stimulating hormone; LH, luteinizing hormone; TRH,thyrotropin- releasing hormone, CRH, corticotropin- releasing hormone; TSH, thyroid- stimulating hormone; VP, vasopressin; (+) indicates stimulatory action and (−) inhibitory action.
Hormones of the posterior pituitary gland
The posterior pituitary gland secretes two hormones into the circulation
Oxytocin is a small peptide hormone that stimulates smooth muscle contraction in the uterus and breast; it functions in parturition (childbirth) and lactation. Vasopressin (VP), also known as antidiuretic hormone (ADH), is a 9-amino acid cyclic peptide with functions linked to the control of water clearance and homeostasis, and is described in detail in Chapter 24. In the human VP, the amino acid in position 8 in the peptide is arginine, and thus it is referred to as arginine-vasopressin (AVP).
Hormonal axes of the hypothalamo–anterior pituitary regulatory system
There are five separate endocrine axes passing through this system (Fig. 39.4)
Three of these form a complex three-level endocrine system, in which the hormones of the pituitary gland, thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH) and luteinizing hormone (LH), may be regarded solely as trophic hormones for other target organs (i.e. thyroid, adrenals, gonads). In these axes, sophisticated control is exercised via a cascade in which each endocrine organ in the axis amplifies both the amount of hormone secreted and the biological half-life of its hormone product compared with the previous organ. The fourth endocrine axis is a hybrid: growth hormone (GH) is both a trophic hormone and has actions in its own right. The fifth and final endocrine axis mediates secretion of prolactin, which is unique in that it is not a trophic hormone. Distinct clinical syndromes are produced by either deficiency or excess of any of the anterior pituitary hormones except prolactin, deficiency of which does not lead to clinically significant problems in humans (Table 39.5).
Table 39.5
Clinical conditions associated with pituitary hormone disorders
|
Hormone |
Deficiency |
Excess |
|
TSH |
Hypothyroidism |
Thyrotoxicosis |
|
ACTH |
Hypoadrenalism |
Cushing's disease |
|
FSH/LH |
Hypogonadism |
Precocious puberty |
|
GH |
Short stature |
Gigantism/acromegaly |
|
Prolactin |
None |
Galactorrhea/infertility |
The hypothalamo–pituitary–thyroid axis
Thyrotropin-releasing hormone (TRH)
TRH is manufactured in the hypothalamus and transported via the portal circulation to the pituitary where it ultimately stimulates secretion of TSH
TRH is a modified tripeptide released in pulsatile fashion by peptidergic hypothalamic nuclei and transported to the anterior pituitary by the portal circulation. TRH stimulates TSH synthesis and secretion by binding to G-protein coupled receptors on the pituitary thyrotroph cell membrane that are linked to phospholipase C. The resulting increase in intracellular inositol trisphosphate (IP3) stimulates the release of calcium from intracellular storage sites and so leads to secretion of preformed TSH. More chronic actions of TRH include stimulation of TSH subunit biosynthesis and TSH glycosylation. The number of TRH receptors on the thyrotrophs is downregulated both by the concentration of TRH itself and by thyroid hormones.
Thyroid-stimulating hormone (TSH)
TSH, or thyrotropin, is a small glycoprotein synthesized by pituitary thyrotrophs
TSH consists of two noncovalently linked subunits and contains about 15% carbohydrate. The α-chain is identical to that found in other glycoprotein hormones (LH, FSH, βHCG) and so the specificity is conferred by the β-chain. The synthesis of each subunit is directed by separate messenger ribonucleic acids (mRNAs) encoded by separate genes on different chromosomes. The carbohydrate side chains are complex mixtures of unmodified, acetylated, and sulfated sugars. TSH is secreted in a pattern that is both pulsatile and circadian, and it has a plasma half-life of about 65 minutes.
TSH acts on the thyroid gland and influences virtually every aspect of thyroid hormone biosynthesis and secretion
TSH, like TRH, acts via a specific G-protein coupled receptor. However, in this case the receptor is expressed on follicular cells in the thyroid gland, and it is coupled to adenylyl cyclase and thus protein kinase A (or cAMP-dependent protein kinase). cAMP-dependent protein kinases control virtually every aspect of thyroid hormone biosynthesis and secretion, including iodide transport, iodothyronine formation, thyroglobulin proteolysis, and thyroxine de-iodination. TSH also stimulates growth of the thyroid gland. Indeed, activating somatic mutations in the TSH receptor are thought to account for many cases of multinodular goiter, a very common irregular enlargement of the thyroid gland, particularly when associated with excess secretion of thyroid hormone.
Negative feedback by thyroid hormones occurs at both hypothalamic and pituitary levels. At the pituitary level, thyroxine (T4) and tri-iodothyronine (T3) inhibit TSH secretion by decreasing both the biosynthesis and release of TSH through regulation of gene transcription and TSH glycosylation. T3, the biologically active form of the hormone, is a more potent feedback inhibitor than T4 and indeed, much of the feedback inhibition by T4 requires its conversion intracellularly to T3 by deiodinase type 2.
Thyroxine and tri-iodothyronine
T4 is produced exclusively in the thyroid gland and is more abundant than T3, which is the biologically active form
Thyroxine (T4, also known as tetra-iodothyronine) and T3 are structurally simple molecules, being iodinated thyronines produced by the coupling of a phenyl group detached from one tyrosine to the phenyl group of a second intact tyrosine (Fig. 39.5). The biosynthesis of T4 and T3 occurs on the surface of thyroglobulin, a large tyrosine-rich glycoprotein that accounts for about 75% of the protein content of the thyroid gland. Iodination of thyroglobulin occurs after it has been secreted into the follicular lumen, and secretion of T4 and T3 requires enzymatic hydrolysis of thyroglobulin, which is located in the follicular colloid. During hydrolysis and subsequent de-iodination, the released iodide is conserved and reutilized (Fig. 39.6).

FIG. 39.5 Structures of the thyroid hormones thyroxine (T4), tri-iodothyronine (T3 ) and reverse T3 (rT3).

FIG. 39.6 Mechanism of biosynthesis of thyroid hormones.
(1) Iodide is concentrated in follicular epithelial cells after entry via a sodium iodide symporter, before (2) oxidation by thyroid peroxidase (TPO) in the peroxisome to iodine. (3) At the plasma membrane adjacent to the follicular lumen, conversion of tyrosyl (Y) residues on the surface of thyroglobulin to either mono-iodotyrosine (MIT) or di-iodotyrosine (DIT) occurs. Coupling of iodinated tyrosines to form either T4 (DIT + DIT) or T3 (MIT + DIT) then occurs. (5)Thyroglobulin is endocytosed and (6) hydrolyzed in lysosomes to release free T3 and free T4. (7) The thyroid hormones are transported to the plasma membrane and released into the bloodstream by mechanisms which are not yet clear. TSH influences virtually every step in thyroid hormone synthesis and release through the cAMP/protein kinase A pathway.
Thyroid hormone bioactivity is regulated by controlling the conversion of T4 into T3 by deiodination; this is mediated by three deiodinase enzymes.
80–95% of the thyroid hormone secreted by the thyroid gland is T4, with T3 as the minor component
However, T3, produced by 5′ deiodination of T4, is the biologically active form of the hormone, and T4 may thus be regarded as a prohormone. 5′ deiodination may occur in the thyroid gland, in target tissues or in other peripheral tissues, and is accomplished by iodothyronine deiodinases type 1 and 2 (DI1 and DI2). The type 2 enzyme is particularly important in controlling nuclear T3 levels, while the physiologic role of the kinetically inefficient type 1 deiodinase is less clear at present. The type 3 enzyme is the major catabolic enzyme; it catalyzes the removal of an iodide from the 3 rather than 5′ position, resulting in reverse T3 (rT3), which is inactive, and similarly de-iodinates and inactivates T3. Thus, control of the de-iodination of T4 is one method of controlling thyroid hormone bio-activity. About 80% of T4 is metabolized by de-iodination, with about equal amounts of T3 and rT3 being produced. The remaining T4 is conjugated with sulfate or glucuronic acid and deactivated by deamidation or decarboxylation. In severe illness there is good evidence that activation of T4 to T3 by DI1 and DI2 is impaired, while the expression of DI3 is increased. This results in low T3 levels, which is an early feature of the so-called ‘sick euthyroid syndrome’.
The biologically active fraction of T3 and T4 in plasma (free T3 or free T4, i.e. a fraction which is not bound to protein), represents, in each case, less than 1% of the total concentration of the hormone
The daily production of T4 is approximately 10% of the extrathyroidal pool, much of which is bound to thyroid hormone binding globulin (TBG) or albumin in plasma. Approximately 80% of this T4 is converted to T3 by extrathyroidal deiodination. The turnover of T3 is much greater than that of T4. The biologically active component of T4 and T3 in plasma is the free fraction – that not bound to proteins. This fraction, which is a measure of thyroid hormone status, represents less than 1% of the total T4 and T3. Most clinical laboratories now measure free hormone levels specifically, to avoid problems in interpretation of total hormone levels caused by fluctuations in binding proteins.
![]() Advanced concept box Membrane transporters for thyroid hormone
Advanced concept box Membrane transporters for thyroid hormone
Traditionally it was thought that lipophilic hormones such as steroids and thyroid hormones simply diffused across lipid bilayers to reach their intracellular receptors. However, in the last few years it has been shown that the plasma membrane MCT8 protein functions as a thyroid hormone transporter, and that genetic deficiency of the protein leads to severe psychomotor retardation, and high T3/T4 ratios. Affected patients do not have peripheral features of hypothyroidism, however. Since this discovery several different thyroid hormone transporters have been identified, and whether other lipophilic hormones also have similar, specific mechanisms governing cell entry remains an interesting question.
Biochemical actions of thyroid hormones
Thyroid hormones may be considered the accelerator pedal of metabolism
T3 increases the metabolic rate of a wide range of tissues and thereby increases the whole-body basal metabolic rate. Most of these actions result from binding of T3 to its nuclear receptors α and β, which are encoded by separate genes (Chapter 40), and alteration in transcription rates of genes in target tissues. Among the many cellular changes which result from this altered program of gene expression, it has been shown that thyroid hormones increase ATP utilization by increasing sodium-potassium (Na+/K+)-dependent ATPase activity, and increase mitochondrial oxidative metabolism, in part by direct upregulation of key mitochondrial biogenesis factors. Adipose tissue lipolysis is also stimulated by cAMP-dependent activation of hormone-sensitive lipase, thus producing fatty acids that can be oxidized to generate the ATP used for thermogenesis. Increase in both glycogenolysis and gluconeogenesis occurs to balance the increased use of glucose as a fuel for thermogenesis. The rate of synthesis of many structural proteins, enzymes, and other hormones is also affected as a result of thyroid hormone action, and microarray-based transcriptomics (Chapter 36) can now be used to define the many hundreds or thousands of genes whose expression is altered by activation of the thyroid hormone receptor.
![]() Advanced concept box A tale of two thyroid hormone resistance syndromes
Advanced concept box A tale of two thyroid hormone resistance syndromes
Thyroid hormone exerts its cellular effects through changes in gene expression mediated by two different nuclear receptors, thyroid hormone receptors α and β (TRα and TRβ). TRα is the dominant isoform in bone, GI tract, and in cardiac and skeletal muscle, while TRβ predominates in the liver, kidney and parts of the hypothalamus. In keeping with this, human genetic defects in both TRα and TRβ have been described. TRβ defects are much more common and cause elevated thyroid hormone levels with detectable TSH. The very recently described TRα defects, in contrast, produce delayed skeletal maturation, constipation and mild mental retardation associated withnormal TSH and high T3/T4 ratio (see Bochukova et al. A mutation in the thyroid hormone receptor α gene. N Engl J Med. 2012;19:366:243–239 and Beck-Peccoz P. Syndromes of thyroid hormone resistance. Ann Endocrinol (Paris). 2005;66:264–269.
Clinical disorders of thyroid function
Thyroid disease is common, affecting almost 3% of the population; nine times as many women as men are affected
More than 95% of thyroid disease originates in the thyroid gland and much of this is autoimmune in origin. Antibodies may arise against several components of thyroid cells, including the peroxidase-rich microsomes. Lymphocyte infiltration and progressive destruction of the thyroid gland ensue, leading to hypothyroidism, the commonest type of thyroid dysfunction. Some thyroid autoantibodies bind to the TSH receptor. If the autoantibodies bind to but do not stimulate the gland, the thyroid hormone production falls and the patient becomes hypothyroid, with increased plasma TSH and reduced free T4. If the autoantibodies bind and stimulate, however, they mimic the effect of TSH, breaking the normal negative feedback loop and producing thyroid hormone oversecretion, and the patient will be hyperthyroid (thyrotoxic) with increased plasma free T4 and suppressed TSH.
Hypothalamic and pituitary causes of hypothyroidism are often the result of impaired TSH secretion secondary to pressure from an adjacent tumor. Pituitary TSH-secreting tumors are an extremely rare cause of hyperthyroidism, and often produce abnormally high ratios of TSH α-subunit to β-subunit, and abnormal glycosylation patterns of TSH which may be useful in diagnosis.
![]() Clinical box A 60-year-old woman with weight gain, intolerance of cold, and tiredness: hypothyroidism
Clinical box A 60-year-old woman with weight gain, intolerance of cold, and tiredness: hypothyroidism
A 60-year-old woman comes to the clinic and complains of weight gain, intolerance of cold, and tiredness. She also says that she has recently become less alert mentally, but attributes this to aging. On questioning she admits that she has also suffered severe constipation of late. She says that two members of her family had ‘thyroid trouble’. On examination, she is moderately obese, has dry, cool skin, a puffy face, and a slow heart rate of 50 beats per minute. The thyroid gland is not palpable. Her free thyroxine (T4) was 5 pmol/L (0.4 ng/dL) (reference range 9–25 pmol/L (0.7-2.0 ng/dL)) and TSH was 60 mU/L (reference range 0.4–4 mU/L).
Comment.
Symptoms of hypothyroidism at an early stage can be fairly nonspecific. The best laboratory test for the diagnosis of hypothyroidism is the measurement of the plasma TSH concentration. The elevated level of TSH suggests a primary thyroid disorder. Subsequently this lady's blood was shown to be positive for anti-microsomal and anti-thyroglobulin antibodies. A diagnosis of lymphocytic thyroiditis (Hashimoto's thyroiditis) was made. She was treated with thyroxine.
![]() Clinical box A 35-year-old woman complaining of palpitations and fatigue: hyperthyroidism
Clinical box A 35-year-old woman complaining of palpitations and fatigue: hyperthyroidism
A 35-year-old woman came to her physician complaining of palpitations, difficulty climbing stairs and general fatigue. She also said that she had lost 4 kg of weight recently despite a good appetite and no attempt at dieting. She also reported occasional diarrhea, and increasingly infrequent and light menstrual bleeds.
On examination, her skin was warm and moist and she had a fine tremor of outstretched hands. There was mild weakness of the thigh muscles. She had tachycardia (110/min). She also had a mild thyroid enlargement (goiter) and a bruit over the gland. Thyroid function tests show suppressed TSH level (<0.05 mU/L; reference range 0.4–4 mU/L) and increased thyroxine (T4 = 41 pmol/L (3.3 ng/dL); reference range 9–25 pmol/L (0.7-2.0 ng/dL)) and tri-iodothyronine (T3 = 17 pmol/L (1105 pg/dL); reference range 3.5–6.5 pmol/L (228-422 pg/dL)). Anti-thyroid receptor antibodies were detected.
Comment.
In hyperthyroidism, the TSH level is suppressed by high circulating thyroid hormones. The low TSH level and high thyroid hormone concentrations suggest hyperthyroidism. The presence of thyroid receptor antibodies confirms that the cause is Graves' disease, an autoimmune thyroid disorder.
![]() Advanced concept box The GnRH pulse generator and human genetics
Advanced concept box The GnRH pulse generator and human genetics
For the first few months of life the hypothalamic-pituitary-gonadal axis is active before it is shut down, going into a quiescent state until it is reactivated at puberty. There are several lines of evidence suggesting that this is largely determined centrally, and a poorly defined group of hypothalamic neurons known as the GnRH ‘pulse generator’ are the main focus of interest. Human genetic studies over the past decade combined with study of genetically modified mice have, however, yielded some key insights, and it is now known that the neuropeptides kisspeptin and neurokinin B are critically required for normal maturation of GnRH secretion, with genetic defects in the genes encoding either kisspeptin, neurokinin B or either of their receptors leading to absent or severely impaired maturation. Much work is now being devoted to understanding the behavior of a group of neurons that are closely apposed to GnRH neurons, and that co-express kisspeptin, neurokinin and also dynorphin, as these neurones have connections and properties that suggest they may form an integral part of the elusive GnRH pulse generator.
The hypothalamo–pituitary–adrenal axis
Corticotropin-releasing hormone (CRH)
CRH is a 41-amino acid peptide secreted by the paraventricular nucleus (PVN)
CRH acts via a G-protein coupled receptor on pituitary corticotroph cells via the cAMP second messenger system to stimulate both synthesis and secretion of ACTH. A second hormone from the PVN, vasopressin (VP), potentiates the response of the pituitary to CRH, in part by increasing the amount and extent of expression of the CRH receptor. Negative feedback by cortisol inhibits both CRH and VP secretion, as well as reducing responsiveness of corticotrophs to stimulation.
Adrenocorticotropic hormone (ACTH)
ACTH is synthesized as a 241-amino acid precursor molecule, pro-opiomelanocortin (POMC)
POMC is cleaved at multiple sites to release several hormonally active peptides, including the endorphins and melanocyte-stimulating hormones. In addition to the pituitary, POMC may also be produced in large quantities by certain malignancies, giving rise to ectopic ACTH syndrome.
ACTH itself is composed of 39 amino acids with the biological activity residing in the N-terminal 24 residues
ACTH is secreted in stress-related bursts superimposed on a marked diurnal rhythm, with a rapid surge in production from around 03.00 h and a peak plasma concentration at about 05.00 h. It is transported unbound in plasma and has a half-life of about 10 minutes. ACTH stimulates the synthesis and release of glucocorticoid hormones by interacting with cell surface G-protein coupled receptors on the adrenal cortex that stimulate cAMP production. Acute increases in the adrenal synthesis of cortisol occur within 3 minutes, principally by stimulating the activity of cholesterol esterase. Longer-term effects of ACTH include induction of transcription of the genes that encode steroidogenic enzymes, and it is also required to maintain thezona fasciculata and zona reticularis of the gland, meaning that long-term lack of ACTH eventually abolishes the ability of the gland to respond to an acute challenge by increasing cortisol and androgen synthesis (Chapter 17).
Negative feedback by cortisol occurs within two timeframes, acting at both the hypothalamic and pituitary levels. Fast feedback alters the release of hypothalamic CRH and the CRH-mediated secretion of ACTH. Slow feedback results from reduced synthesis of CRH plus suppression of POMC gene transcription, which results in reduced ACTH synthesis.
Biosynthesis of cortisol
Cortisol is the major glucocorticoid synthesized in man in the adrenal cortex, and is under the direct control of pituitary ACTH
A simplified scheme of steroid biosynthesis is shown in Figure 39.7 (see also Fig. 17.12). Cholesterol is the precursor for all steroid hormones. Cleavage of the cholesterol side chain liberates the C-21 corticosteroids; further side chain cleavage yields the C-19 androgens, and aromatization of the A ring results in the C-18 estrogens. The structures of the key steroid hormones are shown in Chapter 17. Several of the steroidogenic enzymes are members of the cytochrome P-450 superfamily of oxidases.

FIG. 39.7 Summary of steroid hormone biosynthesis. (1) cholesterol 20,22-desmolase; (2) 3β-hydroxysteroid dehydrogenase Δα4,5-oxosteroid isomerase; (3) 21-hydroxylase; (4) 11-hydroxylase; (5) aldosterone synthase; (6) 17α-hydroxylase; (7) 17,20-lyase/desmolase; (8) 17β-hydroxysteroid dehydrogenase; (9) aromatase. These enzymes are part of the cytochrome P-450 dependent superfamily of NADPH-dependent monooxygenases (see also Chapter 30; compare Figs 17.11 and 17.12).
The plasma concentration of cortisol shows a pronounced diurnal rhythm, being some 10 times higher at 08.00 h than at 24.00 h. This parallels the marked diurnal rhythm of secretion of ACTH. Approximately 95% of cortisol in plasma is bound to proteins, mainly the cortisol-binding globulin, (CBG). As cortisol concentration rises, the percentage of free cortisol also rises, indicating that CBG binding is saturable. Cortisol has a half-life in plasma of about 100 minutes. It is metabolized in the liver and other organs by a combination of reduction, side chain cleavage, and conjugation to produce a wide range of inactive metabolites that are excreted in urine (see Fig. 17.13).
Cortisol has diverse effects on metabolism, and tissue growth and repair
Cortisol has wide-ranging effects on metabolism, immune function, the cardiovascular system and skeleton, mediated by alterations in expression level of thousands of genes. As the name glucocorticoid suggests, cortisol has a major influence on glucose homeostasis: it is a counterregulatory hormone, working through nuclear receptors to induce the biosynthesis of gluconeogenic enzymes, while at the same time inhibiting glucose uptake and metabolism in peripheral tissues. Glycogen synthesis and deposition are increased, and lipolysis in adipose tissue is stimulated. Protein and RNA synthesis are stimulated in the liver but inhibited in muscle; proteolysis in muscle produces the amino acid substrates for hepatic gluconeogenesis. Cortisol works in tandem with several other hormones including insulin and GH in regulating intermediary metabolism, and these metabolic effects are best understood as part of theendocrine response to physiologic stress (Chapter 21).
Cortisol also modulates the immune system through effects on cytokine production and enhanced leukocyte apoptosis, effects which are exploited pharmacologically in the use of potent glucocorticoids to treat inflammatory conditions. It is, moreover, permissive for the effect of many vasoconstrictors such as catecholamines, has direct effects on the heart and on the kidney, where it enhances water clearance and electrolyte reabsorption, and bone, influencing turnover through a variety of mechanisms.
Clinical disorders of cortisol secretion
Hyposecretion of cortisol may occur as a result of hypothalamic, pituitary or adrenal failure
The diagnosis of cortisol deficiency and its cause relies on the clinical presentation, carefully timed measurement of cortisol and ACTH, and the extent of the cortisol response to synthetic ACTH (Synacthen).
Addison's disease refers specifically to primary adrenal failure in which secretion of all adrenal hormones is decreased, usually because of autoimmune disease or infection (e.g. by tuberculosis or cytomegalovirus). Biochemically, it is characterized by low serum sodium, high serum potassium, acidosis, and impaired cortisol response to synthetic ACTH (Synacthen), together with an elevated plasma ACTH. Cortisol replacement, usually together with a mineralocorticoid, is an effective treatment for this life-threatening condition. Addison's disease can be associated with a markedly elevated TSH which resolves with glucocorticoid therapy. This is important to recognize, as erroneous diagnosis of primary hypothyroidism and treatment with thyroxine may dangerously exacerbate signs and symptoms of hypoadrenalism.
Deficiencies of CRH and ACTH are only rarely isolated and more commonly accompanied by deficiencies of other hypothalamic or pituitary hormones due, for example, to a compressive pituitary tumor. Because glucocorticoids are also involved in normal clearance of free water, their deficiency can mask coexisting ADH deficiency, and replacement leads to dramatic diuresis.
![]() Clinical box A 40-year-old woman who collapsed at home: primary adrenal insufficiency is an endocrine emergency
Clinical box A 40-year-old woman who collapsed at home: primary adrenal insufficiency is an endocrine emergency
A 40-year-old woman was admitted as a medical emergency following a collapse at home 2 days after contracting influenza. For 1 year she had been complaining of chronic weakness and fatigue, and lately of abdominal pain, nausea, vomiting, anorexia and confusion. She was dehydrated and hypotensive with pigmentation of the palmar creases and buccal mucosa. Biochemical analysis revealed Na+ 115 mmol/L (135–145 mmol/L), K+ 5.9 mmol/L (3.5–5.0 mmol/L), urea 12 mmol/L (2.5–6.5 mmol/L) and glucose 2.9 mmol/L (4.0–6.0 mmol/L). Baseline serum cortisol was 55 nmol/L (2 ug/dL), and did not rise adequately 30 min after administration of synthetic ACTH (Synacthen).
Comment.
This is an acute presentation of Addison's disease most likely resulting from progressive autoimmune destruction of the adrenal cortex. The nonspecific symptoms of cortisol deficiency became critical after the stress of a minor illness. The hypotension and electrolyte imbalance result from mineralocorticoid deficiency. The pigmentation is due to elevated POMC-derived peptides such as the melanocyte-stimulating hormones. The diagnosis is made by the impaired cortisol response to synthetic ACTH. Treatment is rehydration with intravenous saline plus intravenous hydrocortisone followed by daily oral hydrocortisone and a mineralocorticoid. Other autoimmune diseases commonly coexist in the patient or their family. Increased frequency of severe hypoglycemia in patients with type 1 diabetes is one presentation to watch for, because of the loss of the glucose counterregulatory effects of cortisol.
Cortisol hyposecretion may also result from a genetic disorder in the steroid biosynthetic pathway: congenital adrenal hyperplasia
The most frequently affected enzyme is the steroid 21-hydroxylase (Fig. 39.7). In nearly complete enzyme deficiency, deficiency of cortisol and aldosterone leads to salt wasting and hyponatremia in the first few days of life, a dramatic presentation which is easy to recognize. Because of the loss of negative feedback by cortisol, ACTH levels rise and try to drive increased amounts of substrate through the enzymatic block at the expense of making an excess of 17-hydroxyprogesterone (17OHP), which is converted to androgens and may masculinize female external genitalia before birth. Partial defects in the enzyme may be compensated for by elevated ACTH levels sufficiently to maintain cortisol levels, though the build-up of metabolites proximal to the defective enzyme in the pathway still occurs. Consequently this presents in teenage girls as hirsutism (growth of male type and distribution of hair in women) and menstrual irregularity. The steroid profile of a patient with congenital adrenal hyperplasia is given in Fig.17.13.
Hypersecretion of cortisol results in Cushing's syndrome – among the most challenging of endocrine disorders
Exogenous glucocorticoids, commonly used to suppress the immune system in a range of inflammatory disorders, are the commonest cause of clinical Cushing's syndrome. However, Cushing's syndrome may also result from disorders of the hypothalamus, pituitary (around 80%) or adrenal gland (around 15%), or it may be the consequence of ectopic ACTH syndrome. Cortisol excess leads to remodeling of adipose tissue with deposition of fat in the face and trunk and loss of fat on the limbs, wasting of skeletal muscle, thinning of skin, and slow healing (Fig. 39.8). Cortisol excess also commonly produces diabetes and hypertension, and usually suppresses the hypothalamic gonadal axis, seen as cessation of menstrual bleeds in women.

FIG. 39.8 Cushing's syndrome due to cortisol hypersecretion.
Livid ‘striae’ are shown, caused by weakening of dermal connective tissue allied to increased abdominal fat tissue.
![]() Clinical box A woman with fatigue, and rapid weight gain cushing's syndrome may present a diagnostic dilemma
Clinical box A woman with fatigue, and rapid weight gain cushing's syndrome may present a diagnostic dilemma
A 45-year-old woman presented with fatigue, rapid weight gain with central obesity, fullness and redness of her face (so called ‘plethora’), and loss of regular menstrual periods. She was mildly hypertensive, and her family doctor had found her also to be diabetic for which she had received dietary advice. Urinary free cortisol was 1000 nmol/24 h (38 ug/dL) (reference range <250 nmol/24 h (<9 ug/dL/24h)); serum cortisol was 500 nmol/L at 24 h (reference range <50 nmol/L) and her 0800 h cortisol was 550 nmol/L (20.9 ug/dL) after 1 mg of dexamethasone (a potent synthetic glucocorticoid) (reference range <50 nmol/L) (<1.8 ug/dL). Plasma ACTH was 100 ng/L (reference range <80 ng/L).
Comment.
This woman has hypercortisolism, known as Cushing's syndrome. The elevated urine cortisol, elevated midnight serum cortisol and failure to suppress following dexamethasone administration support the diagnosis. The ACTH result indicates that the cause is either a pituitary tumor (most likely) or ectopic ACTH secretion from an occult tumor. In this case magnetic resonance imaging (MRI) of the pituitary revealed a clear tumor of 0.5 cm diameter; diagnosis of Cushing's syndrome and localization of the problem are often not this easy! Pituitary surgery is the appropriate definitive treatment in this case.
The diagnosis of Cushing's syndrome may be extremely demanding for several reasons
Because of the pronounced circadian variation in serum cortisol levels, random measurement of cortisol is of little use in the diagnosis of Cushing's syndrome. Instead, 24-hour urinary free cortisol, which provides a measurement of total production of cortisol over 1 day, is a common screening test. An abnormal circadian profile of cortisol secretion, with sustained levels at night when they usually almost undetectable, may produce Cushing's syndrome even if there is little, if any, increase in total daily production of cortisol. This may be detected by sampling of cortisol at 09.00 h and midnight, showing loss of normal circadian rhythm. If this test is undertaken carefully without provoking an endocrine stress response by taking blood, it can be a sensitive means of detecting hypercortisolism.
Endogenous Cushing's syndrome usually arises from autonomously functioning secretory tumors
Some of the diagnostic challenges in Cushing's syndrome arise from the variable behavior of such tumors as they evolve. Sometimes only episodic cortisol hypersecretion is seen, with testing normal in between episodes, and sometimes aberrant receptor expression due to somatic mutations means that high levels of cortisol are secreted in response to other hormones such as gut-derived peptides.
Once these pitfalls have been overcome and cortisol excess demonstrated, the anatomic site of the problem can be investigated using a range of tests which may include ACTH measurements, selective venous catheterization of the pituitary or the adrenal glands, and dexamethasone (a synthetic glucocorticoid) suppression testing. The principle of this suppression testing is that tumors which have become autonomous will not exhibit normal complete suppression of secretory activity by a dose of potent glucocorticoid. Definitive treatment is usually surgical and should be targeted at the primary cause of the condition. However, some medical means of reducing cortisol synthesis are commonly used in preparation for surgery.
The hypothalamo–pituitary–gonadal axis
Gonadotropin-releasing hormone (GnRH)
GnRH is essential for the secretion of intact FSH and LH
The hypothalamus has a major role in the control of gonadal function in both males and females, acting via secretion of GnRH. GnRH is a decapeptide synthesized by varioushypothalamic nuclei and secreted by a relatively small number of neurons into the portal system for transfer to the pituitary. GnRH neurons thus serve as a fairly narrow conduit for transmission of central reproductive signals, and are vulnerable to many types of pathology. GnRH secretion is highly pulsatile and it induces the synthesis and secretion of both FSHand LH. GnRH secretion is held in quiescence between infancy and puberty. Increased frequency and amplitude of hypothalamic GnRH pulses is the first measurable stage of the onset of puberty. Its release is subject to negative feedback by progesterone, prolactin, and sometimes estrogen, among other hormones. GnRH acts through its cell surface receptor to increase intracellular calcium, hydrolyze phospho-inositides, and activate protein kinase C. Long-acting GnRH agonists can also cause downregulation of GnRH receptors and reduced FSH and LH secretion. Such agonists are now used to treat prostate cancer and to prepare infertile women for assisted-conception programs.
![]() Advanced concept box Human growth and longevity
Advanced concept box Human growth and longevity
Although genetic defects in GH or IGF-I action lead to severely impaired growth, studies in many experimental organisms suggest that reduced IGF-I signaling may extend lifespan. Recent studies in Ecuador of individuals affected by Laron-type dwarfism due to GH receptor mutation suggested that this may also be true in humans, as despite their short stature, those with GH receptor defects appeared protected from diabetes, cancer, and had improved biochemical markers of ageing. (See Guevara-Aguirre J et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med 2011;3:70ra13.)
Follicle-stimulating hormone and luteinizing hormone
Although FSH and LH have been given their names on the basis of their function in the female, they are also secreted and functional in the male
Both FSH and LH are secreted in the male and female under the influence of GnRH, and alterations in the GnRH pulse frequency and amplitude can influence the relative amounts of FSH and LH secreted by the gonadotroph. FSH and LH are glycoproteins with an identical α-subunit (also shared with TSH) and a specific β-subunit. The gene for LH has recently undergone duplication to produce βHCG, a gonadotropin secreted by embryonic tissues.
GnRH is essential for the secretion of intact FSH and LH, but feedback from estradiol and testosterone plus gonadal peptides such as inhibin have an important role in reproductive function. Feedback by estradiol is especially interesting because it may have either a negative or a positive effect on gonadotropins, depending on the stage of the menstrual cycle. An outline of the control of the hypothalamo-pituitary-gonadal axes, both for adult men and women, is shown in Figure 39.9. The half-life of LH in plasma is approximately 50 minutes; FSH has a longer half-life of about 4 hours. Both FSH and LH concentrations vary considerably depending on age and sex.

FIG. 39.9 Control of the hypothalamo-pituitary-gonadal axes.
(A) In men, testosterone is produced from cholesterol in the Leydig cell under LH stimulation. Testosterone and FSH support spermatogenesis. (B) In women, estradiol (E2) is produced by the granulosa cell and the developing follicle after feedback stimulation. E2 feedback is mainly negative but, in midcycle, there is a positive E2 feedback resulting in the surge of LH that causes ovulation. Progesterone (P) is secreted by the resultant corpus luteum.
Actions of FSH and LH on the testes
FSH and LH influence spermatogenesis
In the male, testosterone biosynthesis occurs in the Leydig cells of the testes under the primary influence of LH (Fig. 39.10) acting through the G-protein-coupled LH receptor. High intratubular testosterone levels cooperate with FSH in promoting spermatogenesis in the spermatic tubule: FSH binds to its specific receptor on the Sertoli cell of the testes and, by a mechanism similar to LH, induces increased synthesis of several proteins, including androgen-binding protein (ABP) and inhibin. ABP is secreted into the seminiferous tubular lumen where it binds testosterone (or its active form dihydrotestosterone). This ensures a high local androgen concentration that, together with FSH, brings about the meiotic divisions that are necessary for spermatogenesis. Inhibin has a role in the FSH negative feedback loop.

FIG. 39.10 Mechanism of action of testosterone.
Testosterone from the testis enters a target cell and binds to the androgen receptor, either directly or after conversion to 5α-dihydrotestosterone (DHT). DHT binds more tightly than testosterone, and the DHT–receptor complex binds more efficiently to chromatin. Actions mediated by testosterone are shown by purple lines, those mediated by DHT are shown by blue lines.
Biochemical actions of testosterone in the male
Testosterone not only influences gonadotropin regulation and spermatogenesis, but is also a natural anabolic steroid
Nearly 95% of serum testosterone comes from the testes, the remainder originating from the adrenal gland. More than 97% of this circulating testosterone is bound to protein, with equal amounts bound to albumin and to the specific sex hormone-binding globulin (SHBG), which is similar in structure to ABP. Testosterone is metabolized in target tissues todihydrotestosterone (DHT) via 5α-reduction, more than doubling its affinity for the nuclear androgen receptor. In addition to effects on gonadotropin regulation and spermatogenesis, androgens induce the development of the reproductive tract from the Wolffian ducts during male sexual differentiation. They also have anabolic effects, stimulating protein synthesis and an increase in muscle mass (Fig. 39.10).
Clinical disorders of testosterone secretion in males
Testosterone deficiency may originate from a wide range of disorders of the hypothalamus, pituitary or testes
Endocrine failure of the testes may occur due to trauma or inflammation of the testes themselves (e.g. due to mumps infection), or due to failure at the level of the hypothalamus or pituitary. Indeed, the gonadotrophs are among the most sensitive of the anterior pituitary cell types to pituitary damage (e.g. due to growth of an adenoma within the bony constraints of the sella turcica) and consequently gonadal failure is often the earliest manifestation of pituitary disease. Central hypogonadism may also be a response to severe weight loss or physiological stress, when it is best seen as an emergency energy-saving mechanism, and is additionally seen in Cushing's syndrome or chronic use of opioid drugs.
There is a series of genetic disorders which manifest as gonadal failure
Klinefelter's syndrome, caused by acquisition of an additional X chromosome in males (karyotype 47, XXY) presents with gynecomastia (abnormal increase in size of the male breast), eunuchoidism and varying degrees of hypogonadism, and is due to abnormal development of the testes. Serum FSH is elevated, and LH is usually elevated, but testosterone may be subnormal. Kallmann's syndrome, in contrast, is a genetic hypothalamic disorder in children, featuring both deficient GnRH production and commonly absent smell (anosmia) due to defective migration of GnRH-producing (and other) neurones to hypothalamus during intrauterine development. Affected individuals present with delayed puberty and subnormal FSH, LH, and testosterone. Androgen deficiency may lead to muscle and fat redistribution and loss of bone mineral density, and these may be corrected, to some extent, by androgen administration (Table 39.6).
Table 39.6
Causes of hypogonadism
|
Site of defect |
Biochemistry |
Causes |
|
Hypothalamus |
Low GnRH* |
Tumors |
|
Pituitary |
High GnRH* |
Tumors |
|
Gonads |
High GnRH* |
Irradiation |
E2, estradiol
*GnRH is not easily measured clinically.
Androgen excess in males is only seen in precocious puberty
Precocious puberty is a rare condition that may result from early maturation of the normal hypothalamo-pituitary-gonadal axis, gain-of-function mutations in the LH receptor or a receptor for another protein known as kisspeptin (metastin), or as a result of a tumor that is secreting either androgen or HCG.
Actions of FSH and LH on the ovary
In the female, FSH promotes estradiol synthesis leading to follicular maturation, while LH leads to follicle rupture and oocyte release
In the mature female, the GnRH pulse generator engages in a dynamic interplay with ovarian cells in maintaining the hormonally driven menstrual cycle (Figs 39.9 and 39.11). Primordial follicles actually begin hormone-independent growth and maturation many weeks before the start of the menstrual cycle, but only at the start of the cycle, when they have acquired the capacity to respond to FSH, are a few follicles preserved from atresia. Rising FSH concentrations stimulate estradiol synthesis in granulosa cells through induction of aromatase and other enzymes (Fig. 39.7). As estradiol is secreted so FSH falls, and this combination plays an important role in the selection of a dominant follicle for further development, though details of the precise mechanism remain to be fully established. Follicular maturation continues under the influence of rising estradiol concentrations. Increasing estradiol because of positive feedback within the dominant follicle, causes the negative central feedback of estrogen to flip into positive feedback, initiating a surge of LH. This LH binds to receptors on the dominant follicle and, in tandem with steroid hormones and other factors such as prostaglandins, induces completion of the first stage of oocyte meiosis, rupture of the follicle and release of the oocyte some 36 hours later. At this time there is a sharp fall in plasma estradiol, followed by a fall in LH.

FIG. 39.11 Changes in hormone secretion during the normal menstrual cycle.
LH, luteinizing hormone; FSH, follicle-stimulating hormone.
The ruptured follicle transforms into the corpus luteum, which secretes progesterone and lesser amounts of estradiol both to sustain the oocyte and to prepare the estrogen-primed uterine endometrium for implantation of a fertilized ovum and the establishment of early pregnancy. In the absence of fertilization, corpus luteum function declines, and progesterone and estradiol secretion falls. This brings about vascular changes in the endometrium leading to tissue involution and menstruation. The decline in steroid secretion stimulates FSH secretion, setting the stage for initiation of the next cycle.
![]() Clinical box Hormone replacement therapy (HRT)
Clinical box Hormone replacement therapy (HRT)
Women normally reach menopause at 45–55 years of age. The absence of ovarian follicles leads to an absence of estrogen and inhibin secretion, resulting in a marked rise in pituitary FSH. There are vasomotor symptoms of estrogen deficiency (flushing) associated with menopause. Long-term postmenopausal estrogen deficiency is known to increase the rate of bone loss, leading to osteoporosis, and also alters lipoprotein metabolism in a way that increases the risk of cardiovascular disease. HRT with an estrogen preparation was standard practice in most developed countries both to relieve the acute symptoms and to provide prophylaxis against the long-term risks. However, recent studies have suggested that HRT may increase a woman's risk for serious conditions including breast cancer, cardiovascular disease, stroke, and pulmonary embolism. Although HRT did have a modest effect on preventing osteoporosis, alternative bone-targeting drugs are also effective. The current recommendations are that HRT should only be used as short-term therapy for vasomotor symptoms that do not respond to other treatment.
Actions of steroid hormones in the female
In a woman with a normal menstrual cycle, it is the progesterone level that is of diagnostic significance
The concentrations of FSH, LH, estradiol, and progesterone vary considerably during the menstrual cycle. In a normally cycling woman, progesterone is of particular diagnostic utility: a serum concentration of more than around 20 nmol/L in the luteal phase (precise values vary among laboratories) is consistent with ovulation (Fig. 39.11). Aside from their roles in the menstrual cycle and reproduction, both estradiol and progesterone have other effects, acting via specific nuclear receptors in target cells. Estradiol, working in tandem with other hormones such as insulin-like growth factor-I (IGF-I), is responsible for linear growth, breast development and maturation of the urogenital tract and the female body shape. In adult life, both estradiol and progesterone support breast function, and estradiol has an important role in preserving bone mineral density. Progesterone is responsible for the rise in basal body temperature during the luteal phase of the menstrual cycle and decreases in progesterone secretion may contribute to premenstrual changes in mood.
Disorders of steroid secretion in the female
Just as in the male, endocrine disorders producing subnormal sex steroid secretion in the female may result from a wide range of disorders in the hypothalamus, pituitary or ovary
Just as for the testes, endocrine failure of the ovaries may occur due to damage or dysfunction of the ovaries themselves, or due to defects at the level of the hypothalamus or pituitary. Indeed, several pathologies, including pituitary adenomas and Kallmann's syndrome, are common to men and women. However, in women the most common genetic disorder affecting the ovary is Turner's syndrome (karyotype 45X), which has characteristic somatic features including short stature, elevated plasma FSH and prepubertal estradiol, and vestigial or ‘streak’ ovaries. Various other endocrine causes of infertility are shown in Table 39.6, but are largely beyond the scope of this text.
Syndromes of excess ovarian steroid secretion lead to precocious puberty in children and infertility and/or hirsutism in the adult
Precocious puberty may arise from early maturation of the normal hypothalamic gonadal axis, an estradiol- or androgen-secreting cyst or tumor of the ovary or the adrenal gland, congenital adrenal hyperplasia, or severe insulin resistance. In the mature female, hypersecretion of androgen in the polycystic ovary syndrome (PCOS) may result in infertility and/or hirsutism. Although the etiology of PCOS remains uncertain, it shows a strong genetic predisposition and current theories favor a key role for aberrant intraovarian androgen metabolism, in some cases strongly influenced by systemic insulin resistance. The nature of the clinically important cross-talk between insulin action and ovarian androgen production is not yet fully worked out, however.
The growth hormone axis
Growth hormone-releasing hormone (GHRH) and somatostatin
GHRH is a 44-amino acid peptide synthesized as part of a 108-amino acid prohormone in the arcuate and ventro-medial nuclei of the hypothalamus, and in the median eminence
GHRH binds to its receptor on the pituitary somatotroph cell and triggers both the adenylyl cyclase and intracellular calcium–calmodulin systems (Chapter 40) to stimulate GH transcription and secretion from the anterior pituitary. Negative feedback from GH and IGF-I results in both a decrease in GHRH synthesis and secretion and an increase in somatostatin synthesis and secretion.
Somatostatin is found as two isoforms, with 14 and 28 amino acids respectively, both of which are produced from the same 116-amino acid gene product. Somatostatin and its receptors are found throughout the brain and also in other organs, notably the gut. Binding of somatostatin to its receptor is coupled to adenylyl cyclase by an inhibitory guanine nucleotide-binding protein, resulting in a decrease in intracellular cAMP. In the context of growth, somatostatin inhibits the secretion of GH. GHRH and somatostatin are released in separate bursts to provide a very fine level of control of GH release. Somatostatin also inhibits the production or action of many other hormones including TSH, insulin, and glucagon and gastrin. Long-acting analogs of somatostatin are thus effective in the management of GH excess as well as tumors secreting a wide range of other hormones, and as an adjunct to pancreatic surgery to inhibit exocrine secretions from the pancreatic residue during healing.
Growth hormone
The greatest secretion of GH occurs in children and young adults, chiefly during sleep
Although GH can exist in variant forms, the major species is a 22 kDa protein. Nearly two-thirds of GH in the circulation is associated with a 29 kDa binding protein that is identical to the extracellular domain of the GH receptor. This binding protein prolongs the half-life of GH in plasma to about 20 minutes.
The normal human pituitary contains approximately 10 mg of GH; less than 5% of this is released each day. GH is released in bursts with a periodicity of 3–4 hours, and greatest secretory activity occurs during sleep. At the peak of a secretory burst, the plasma GH concentration may be 100-fold greater than baseline; this means that no reference interval can be derived for this hormone, and so meaningful measurement requires either a provocative test or multiple samples over the course of a day. Secretory bursts occur most frequently in children and young adults.
GH has a wide range of actions in regulating growth and intermediary metabolism
It is not surprising, in view of the wide range of actions of GH, that several factors other than GHRH and somatostatin can influence GH secretion. These include other hormones (estradiol) and metabolic fuels (glucose). Several pharmacologic agents are also known to influence GH secretion. These factors act at the higher centers and the hypothalamus to alter the pattern of GHRH and somatostatin secretion.
Binding of GH to its receptor precipitates a complex series of intracellular events that lead to the transcription of many enzymes, hormones and growth factors, including IGF-I
The diversity of action of GH makes it difficult to understand or integrate all of its functions; thus it is convenient to think in two distinct phases (Fig. 39.12). The direct actions of GH are on lipid, carbohydrate, and protein metabolism. During hypoglycemia, GH stimulates lipolysis and induces peripheral resistance to insulin. These effects stimulate the use of fatty acids in peripheral tissues, sparing glucose for use in the brain. Previously, GH therapy was used only for treatment of GH deficiency in children in order to stimulate growth; however, treatment is now often extended into adulthood when it can sometimes improve quality of life. This change in treatment strategy illustrates the complex effects of GH on metabolism. During growth, GH stimulates the uptake of amino acids and their incorporation into protein, especially in muscle. The indirect actions of GH are mediated by IGF-I. These actions promote the proliferation of chondrocytes and the synthesis of cartilage matrix in skeletal tissues. Although GH is most often associated with its role in stimulating linear growth, it clearly has other roles in influencing the relative amount and distribution of fat and muscle tissue. Its anabolic effects are the rationale for its current illicit use in sport.

FIG. 39.12 Biochemical functions of the growth hormone (GH).
These can be divided conveniently into direct actions on lipid and carbohydrate metabolism, and indirect actions on protein synthesis and cell proliferation.
Insulin-like growth factor I
IGF-I is the most GH-dependent of a series of growth factors
IGF-I is a 70-amino acid single-chain basic peptide, which has considerable homology with proinsulin. In response to tropic hormones like GH, IGF-I is produced in many tissues, whose growth is then stimulated; that is, IGF-I acts as a paracrine hormone. The liver is the major source of circulating IGF-I, whose function is primarily feedback inhibition of GH secretion. In plasma and other extracellular fluids, IGF-I is complexed to a series of IGF-binding proteins (IGFBPs) of which IGFBP-3 is the most abundant. IGF-I works through the type 1 IGF receptor, which is structurally similar to the insulin receptor and linked to intracellular tyrosine kinase activity. The relative affinities of insulin and IGF-I for their respective receptors mean that, in normal physiology, there is little cross-stimulation, although in pathologic and pharmacologic situations it is possible for insulin to have some action through the IGF-I receptor, and vice versa. This has implications for states of severe insulin resistance, where local activation of the IGF1 receptor has been suggested to underlie some clinical features such as skin overgrowth (acanthosis nigricans). Conversely, IGF-I has a limited therapeutic role in some rare conditions caused by loss of insulin receptor function.
The plasma reference interval for IGF-I in adults aged 20–60 years is fairly constant. It is much lower in young children but rises dramatically during the period of growth and progression through puberty. IGF-I concentrations fall after the sixth decade of life. Thus, IGF-I appears to be a good marker of integrated GH activity.
![]() Clinical test box Biochemical confirmation of pregnancy
Clinical test box Biochemical confirmation of pregnancy
Biochemical confirmation of pregnancy is now achieved by means of a simple and sensitive test that can give reliable results within 2 weeks of fertilization (i.e. before the next menstrual period). Pregnancy tests may be performed by doctors, nurses or even the patients themselves since they are readily purchased from pharmacies. Pregnancy tests are immunoassays that measure the production of human chorionic gonadotropin (HCG). A specific two-site immunoassay is employed (see Fig. 39.2). One antibody recognizes the α-subunit of HCG, while the other only binds to the 32-amino acid segment of the β-subunit that is unique to HCG (Fig. 39.10). Since nonpregnant women do not normally produce HCG, the detection of even low levels from the first stages of placental development is sufficient to confirm pregnancy.
Clinical disorders of GH secretion
GH deficiency in children is one of several causes of short stature, while GH excess in children leads to gigantism and, in adults, to acromegaly
The absence of a plasma reference range for GH means that GH deficiency may only be diagnosed by studying the dynamics of GH secretion, either during sleep or following a stimulation test. Basal IGF-I or IGFBP-3 measurements may serve as a preliminary screening test. Treatment is by regular injection of recombinant human GH. Adults with a definite cause of GH deficiency (hypopituitarism) are also candidates for GH replacement therapy. A rare genetic cause of short stature is Laron dwarfism in which GH levels are elevated but IGF-I levels are subnormal – a GH receptor defect is responsible.
In most cases, GH excess is caused by a pituitary tumor
GH excess is most commonly due to a GH-secreting pituitary tumor, although GHRH-secreting tumors in the hypothalamus and ectopic GHRH production from pancreatic tumors have been described. In children, GH excess manifests itself as gigantism. In adults, the epiphyses of the long bones have closed and so further linear growth is not possible. Therefore, the adult form, acromegaly, is characterized by a thickening of tissues and overgrowth of the bones in the hands, feet and face (Fig. 39.13). Enlarged lips and tongue with lower jaw overgrowth give rise to a classic facial appearance. Excessive sweating and joint pains are also common symptoms. Unfortunately, the progression of acromegaly is so slow that an average of 9 years elapses between the onset of symptoms and diagnosis. Classically, the diagnosis of GH excess has relied on showing inadequate GH suppression during a standard 75 g oral glucose tolerance test and on elevated IGF-I levels. However, pubescent patients may have paradoxic responses to glucose, and IGF-I levels are normally elevated during this developmental stage. Therefore evidence of a pituitary tumor obtained using magnetic resonance imaging (MRI) is critical in making the proper diagnosis. Surgery is the preferred treatment, although long-acting somatostatin is also effective. More recently, pegvisomant, a GH antagonist rationally designed based on structural features of the hormone–receptor interaction, has also proved useful in refractory cases.

FIG. 39.13 Acromegaly.
Enlarged, somewhat square or ‘spadelike’ hands are shown compared to a normal hand in the middle.
The prolactin axis
Dopamine
Dopamine is an inhibitor of prolactin secretion
Prolactin is unique among the pituitary hormones in that it is under predominantly inhibitory control from the hypothalamus (Fig. 39.4). Furthermore, the controlling agent is the simple molecule dopamine (Chapter 41.1), which is produced by tuberoinfundibular dopamine neurons. Dopamine works by stimulating the pituitary lactotroph D2 receptor to inhibit adenylyl cyclase and consequently inhibits both prolactin synthesis and secretion. Several neuropeptides, including TRH, have prolactin-releasing properties, but there is little evidence for a physiologic role.
Prolactin
Prolactin may assist breast growth and milk formation, in association with other pregnancy-related hormones
Prolactin is a 23 kDa protein which is homologous to GH. The primary role for prolactin in humans occurs during pregnancy when prolactin binds to its receptor in mammary tissue and stimulates the synthesis of several milk proteins, including lactalbumin. In other animals, prolactin also has gonadotropic, immunologic, and hematologic effects: it is required for maintenance of pregnancy in rodents, is mitogenic for some immune cells, and it ameliorates some forms of anemia. Prolactin exerts its effects on female reproductive function at multiple levels. Its actions include blocking the action of FSH on follicular estrogen secretion and enhancing progesterone levels by inhibiting steroid-metabolizing enzymes.
![]() Clinical test box Macroprolactin
Clinical test box Macroprolactin
Although antibody-based immunoassays are extremely powerful tools in endocrine diagnosis, they are subject to interference by endogenous antibodies in several ways.One way in which endogenous antibodies can produce confusing results is by binding and aggregating the target hormone in high molecular weight (HMW) complexes. This may be seen for several peptide hormones, but is particularly problematic for prolactin, where so-called ‘macroprolactin’ may account for up to 10% of cases of apparently elevated serum prolactin. This is a problem, as macroprolactin is generally not biologically active, so its detection could lead to unnecessary treatment. There are several ways around this. Most simply, as different immunoassays often have different capabilities to detect macroprolactin, use of more than one assay may provide a strong clue to its presence. Second, a common technique is to precipitate HMW complexes with polyethylene glycol (PEG), with immunoassay both before and after to establish how much signal is lost in the HMW fraction. More accurate quantification may require laborious techniques such as gel filtration.
Clinical disorders of prolactin secretion
There are no known prolactin deficiency syndromes but hyperprolactinemia is very common
Hyperprolactinemia may result from a prolactin-secreting pituitary tumor (prolactinoma), a deficient supply of dopamine from the hypothalamus, or the use of any of a wide range of antidopaminergic drugs. In women, the presenting features of hyperprolactinemia include menstrual irregularity and galactorrhea (discharge of milk from the breast).
A grossly elevated serum prolactin is usually diagnostic of a prolactinoma
For subjects with modest hyperprolactinemia, who are not taking antidopaminergic drugs, the diagnosis is more difficult; pituitary imaging and/or dynamic tests of prolactin secretion will assist the diagnosis of a microprolactinoma. Treatment options include long-acting dopamine agonist drugs or surgery. Prolactinomas need to be observed particularly carefully during pregnancy, when there is physiologic hyperplasia of pituitary lactotrophs in anticipation of lactation.
In men, hyperprolactinemia can cause impotence and prostatic hyperplasia. However, because other conditions more commonly produce these symptoms, the diagnosis of hyperprolactinemia is often missed until prolactinomas become very large and present as hypopituitarism with visual field defects. The latter symptoms arise when the tumor expands out of the pituitary fossa and impinges on the optic chiasm. Such tumors often shrink and fibrose with dopamine agonist therapy. For this reason it is critically important to identify which patients with large pituitary tumors have very high prolactin levels, as they may be able to avoid surgery.
Endocrine systems not considered in this chapter
The human body contains several other endocrine systems not considered in this chapter, though they are governed by the same general principles. Some of these systems are considered in other chapters as part of the physiologic function that they control. Thus the reader is referred to Chapter 21 for carbohydrate homeostasis, Chapter 26 for calcium homeostasis andChapter 24 for water and electrolyte homeostasis and the control of blood pressure. The intracellular signaling systems through which hormones exert their effects are described inChapter 40.
Summary
![]() The endocrine system produces hormones, a structurally diverse group of chemical messengers that regulate and coordinate whole-body metabolism, growth, reproduction, and responses to external stimuli.
The endocrine system produces hormones, a structurally diverse group of chemical messengers that regulate and coordinate whole-body metabolism, growth, reproduction, and responses to external stimuli.
![]() The hypothalamic-anterior pituitary axis is a critical link between the brain and peripheral endocrine glands, and integrates diverse internal and external stimuli into only a few hormonal signals: it controls the synthesis and action of thyroid hormone, glucocorticoids, sex steroids, growth hormone, and prolactin.
The hypothalamic-anterior pituitary axis is a critical link between the brain and peripheral endocrine glands, and integrates diverse internal and external stimuli into only a few hormonal signals: it controls the synthesis and action of thyroid hormone, glucocorticoids, sex steroids, growth hormone, and prolactin.
![]() The hypothalamic-posterior pituitary axis controls the production and release of oxytocin and AVP/ADH. The action of these hormones on cells is controlled by receptors located either on cell membranes or intracellularly.
The hypothalamic-posterior pituitary axis controls the production and release of oxytocin and AVP/ADH. The action of these hormones on cells is controlled by receptors located either on cell membranes or intracellularly.
![]() Feedback mechanisms are important in controlling endocrine systems, and both overactivity and underactivity of these axes produce distinct clinical syndromes.
Feedback mechanisms are important in controlling endocrine systems, and both overactivity and underactivity of these axes produce distinct clinical syndromes.
![]() Laboratory diagnosis of endocrine disorders relies heavily on measurement of hormones in blood; however, careful attention must often be paid to the timing of sampling, and to the nature of the sample required, before meaningful interpretation of the results is possible.
Laboratory diagnosis of endocrine disorders relies heavily on measurement of hormones in blood; however, careful attention must often be paid to the timing of sampling, and to the nature of the sample required, before meaningful interpretation of the results is possible.
Active Learning
1. Trace the two-way flow of information between hypothalamus and ovarian follicles early in the menstrual cycle.
2. How do GH, cortisol, and insulin interact to regulate lipid and carbohydrate metabolism?
3. Because dietary iodide can vary greatly, thyroid hormones need to be synthesized and stored when iodide is available, but thyroid hormones are hydrophobic and cannot be contained within membrane vesicles. How does the thyroid gland solve this problem?
Further reading
Anderson, MS. Autoimmune endocrine disease. Curr Opin Immunol. 2002; 14:760–764.
Cooper, DS. Hyperthyroidism. Lancet. 2003; 363:459–468.
Cooper, DS, Biondi, B. Subclinical thyroid disease. Lancet. 2012; 379:1142–1154.
Dattani, M, Preece, M. Growth hormone deficiency and related disorders: insights into causation, diagnosis and treatment. Lancet. 2004; 363:1977–1987.
Dayan, CM. Interpretation of thyroid function tests. Lancet. 2001; 357:619–624.
Franklyn, JA, Boelaer, K. Thyrotoxicosis. Lancet. 2012; 379:1155–1166.
Henderson, J. Ernest Starling and ‘hormones’: an historical commentary. J Endocrinol. 2005; 184:5–10.
Løvås, K, Husebye, ES. Addison's disease. Lancet. 2005; 365:2058–2061.
Melmed, S. Acromegaly. N Engl J Med. 2006; 355:2558–2573.
Newell Price, J, Bertagna, X, Grossman, A, et al. Cushing's syndrome. Lancet. 2006; 367:1605–1617.
Roberts, CGP, Ladenson, PW. Hypothyroidism. Lancet. 2004; 362:793–803.
Semple, RK, Topaloglu, AK. The recent genetics of hypogonadotrophic hypogonadism – novel insights and new questions. Clin Endocrinol (Oxf). 2010; 72:427–435.
Plant, TM. Hypothalamic control of the pituitary-gonadal axis in higher primates: key advances over the last two decades. J Neuroendocrinol. 2008; 20:719–726.
Schlechte, JA. Prolactinoma. N Eng J Med. 2003; 349:2035–2041.
Schneider, HJ, Aimaretti, G, Kreitschmann-Andermahr, I, et al. Hypopituitarism. Lancet. 2007; 369:1461–1470.
Zimmermann, MB, Jooste, PL, Pandav, CS. Iodine-deficiency disorders. Lancet. 2008; 372:1152–1168.
Websites and downloads
The Endocrine Societ. www.endo-society.org
Endotext.otg. The endocrine source. www.endotext.org.
Thyroid disease manager. www.thyroidmanager.org.
Pituitary Network Associatio. www.pituitary.org
Cushing's Support and Research Associatio. www.CSRF.net
National Endocrine and Metabolic Diseases Information Servic. www.endocrine.niddk.nih.gov/