James A. Fagin
Thyroid follicular cells can give rise to tumors with distinct pathologic and clinical features, as discussed in Chapter 21. Benign and malignant thyroid neoplasms consist of clonal cell populations (1,2,3). This points to their derivation from single precursor cells, with the initial step in tumor development involving the acquisition of a somatic mutation that alters the structure of a gene that confers cells with a growth advantage. Many of the genes involved in the pathogenesis of thyroid tumors have been identified, and found to be preferentially associated with certain tumor types. Cell proliferation is stimulated by extracellular growth factors that activate specific signaling cascades that dictate the orderly sequence of events needed for DNA synthesis and cell division. Molecules acting at multiple steps along growth signaling pathways can function as oncogenes when their structure is disrupted. In addition, loss-of-function mutations of genes coding for growth-inhibitory proteins involved in cell cycle checkpoints or cell survival contribute significantly to tumorigenesis. In the first section of this chapter, the contribution of growth and survival molecules to thyroid cancer pathogenesis based on their cellular function will be discussed.
GROWTH FACTORS AND EICOSANOIDS AS THYROID TUMOR PROGRESSION FACTORS AND PROMOTERS OF ANGIOGENESIS
The abundance of thyroid-stimulating hormone (TSH, or thyrotropin) in plasma is an important determinant of the mitotic rate of thyroid cells (4). High TSH levels promote progression of certain thyroid neoplasms, including many thyroid cancers. By contrast, the role of growth factors present in the tumor environment is not as well defined. Whereas growth of normal cells is highly dependent on external mitogenic stimulation by growth factors, a classical feature of cancer cells is that they are to a large extent released from this constraint. Nevertheless, tumor cells retain variable degrees of responsiveness to polypeptide growth factors, and these may contribute to tumor progression or development. Fibroblast growth factors (FGF) 1 and 2 are mitogens for thyroid follicular cells (5), and are overexpressed in multinodular goiters as well as benign and malignant thyroid neoplasms (6,7,8). TSH requires the presence of insulin-like growth factor-I (IGF-I) to exert its full mitogenic effect in human thyroid cells. Moreover, overexpression of IGF-I and the IGF-I receptor in thyroid cells of transgenic mice induces goiter formation (9). Both IGF-I and IGF-II are overexpressed in human thyroid cancers (10,11). Whereas IGF-I is primarily produced by cells in the tumor stroma (12), thyroid cancer cells are believed to be the source of local IGF-II production (10). Transforming growth factor-α (TGF-α, and its close relative epidermal growth factor, EGF) is also a mitogen for thyroid follicular cells in vitro, and is locally expressed within the thyroid (13,14). The mechanism by which tumor cells overexpress growth factors does not involve primary modifications of the growth factor genes themselves. Rather, growth factor overexpression is secondary to oncogenic activation of other signaling pathways. For example, mutations of RAS result in increased TGF-α gene expression in rat thyroid FRTL-5 cells (15), and of IGF-I in human thyroid cells (16).
For cancer clones to progress, tumor cells promote growth of capillaries and larger vessels from adjacent tissue in order to deliver nutrients and oxygen and eliminate metabolic waste. Some of the most prominent signals driving the process of angiogenesis have been identified in other cancer types (17,18), and are likely to play similar roles in the thyroid. Vascular endothelial cell growth factors (VEGFs) comprise a family of at least five structurally related polypeptides—VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PIGF)—that modulate blood and lymphatic vessel growth (19). VEGF-A is expressed at higher levels in thyroid cancers than in normal thyroid cells or benign thyroid tumors (20,21,22). VEGF-C, which primarily stimulates lymphangiogenesis, is also overexpressed in thyroid cancers, although this alone is unlikely to account for the predisposition to undergo lymphatic spread, since not all thyroid tumor types prone to lymph node metastasis overexpress this growth factor (23). Besides its mitogenic effects on thyroid cells, FGF-2 is also a potent angiogenic factor (24). TGF-β, which despite inhibiting thyroid cell growth and differentiated function (25) has been implicated in pathogenesis of multinodular goiters (26,27), could play a role in vascular remodeling. It is important to consider the role of stromal-derived growth factors in tumor evolution, as illustrated by recent evidence indicating that modulation of TGF-β signaling in stromal fibroblasts can impact the oncogenic potential of adjacent epithelial cells (28). In addition to polypeptide growth factors, other secreted products present in the tumor microenvironment can promote neovascularization. Thyroid cancers, like other epithelial malignancies, have higher expression of cyclooxygenase-2 (COX-2), the enzyme catalyzing the initial step in prostanoid biosynthesis from arachidonic acid (29,30,31,32). Tumor-derived prostanoid production is a significant contributor to colorectal tumorigenesis in animals and humans (33,34,35,36). Prostaglandin E2 (PGE2), the major product of COX-2 acting via microsomal prostaglandin E synthase (mPGES), has been implicated as a key mediator of these effects. Among other properties, PGE2 is a powerful stimulus for angiogenesis. Recent evidence shows that expression of Ret or Ras mutants induces PGE2 production via increased expression of COX-2 and mPGES1 in rat thyroid cells, thus providing a link between oncogenic tumor initiation, activation of prostaglandin biosynthesis, and tumor neovascularization (37).
DEFECTS IN SIGNAL RECEPTION FROM EXTRACELLULAR LIGANDS
Tyrosine Kinase Receptors
Polypeptide growth factor receptors are common targets of oncogenesis. This can occur through gene amplification of a tyrosine kinase receptor of normal structure, resulting in an increase in the total number of receptors in the plasma membrane. For example, the epidermal growth factor receptor (EGFR, or Erb1) is overexpressed in certain epithelial malignancies and in glioblastomas, and its close relative Erb-B2/neu is amplified and overexpressed in breast and ovarian cancer (38,39,40). Even though in most cases these receptors are structurally intact, their increased abundance favors a higher amplitude of signaling through effector pathways that can favor cell transformation (41). So far, gene amplification of growth factor receptors has not proven to be a common feature of thyroid neoplasms. However, all members of the EGFR family have been reported to be overexpressed in papillary thyroid cancers (42,43). Overexpression of Met, the receptor for hepatocyte growth factor (HGF), is also highly prevalent in these cancers (44,45). This is not due to amplification of the MET or EGFRgenes, but may instead be secondary to induction of gene expression, possibly as a result of illegitimate signaling from mutant RAS or RET/PTC oncogenes (46,47). The predicted increase in responsiveness to the respective ligands for these tyrosine kinase receptors may thus participate in the tumorigenic process.
Growth factor receptors can also lead to cell transformation when their structure is modified through activating mutations. The most significant example relevant to the pathogenesis of papillary thyroid carcinomas are the RET/ PTC oncogenes (48), mutant forms of the tyrosine kinase receptor RET. The RETgene encodes a transmembrane tyrosine kinase receptor whose expression is normally restricted to a subset of neuronal and neuroendocrine cells, and to the collecting duct of the kidney. Ret is a receptor for growth factors belonging to the glial cell–derived neurotropic growth factor (GDNF) family, which also includes neurturin, persephin, and artemin (49). The Ret protein is not normally present in thyroid follicular cells. Chromosomal rearrangements linking the promoter and N-terminal domains of unrelated genes to the C-terminal fragment of RET result in the aberrant production of a chimeric form of the receptor in thyroid cells. Rearrangements of RET resulting in its constitutive activation are believed to play a causative role in the pathogenesis of a significant proportion of papillary carcinomas of the thyroid (PTC) (50). Several forms have been identified that differ according to the 5′ partner gene involved in the rearrangement. RET/ PTC1 is formed by an intrachromosomal inversion of the long arm of chromosome 10, leading to fusion of RET with a gene named H4/D10S170 (51). RET/PTC2 is formed by a reciprocal translocation between chromosomes 10 and 17, resulting in the juxtaposition of the TK domain of RET with a portion of the regulatory subunit of PKAR1A (52). RET/PTC3 is also a result of an intrachromosomal rearrangement and is formed by fusion with the RFG/ELE1 gene (53,54) (Fig. 70B.1). Recently, several additional variants of RET/PTC have been observed in papillary carcinomas from individuals exposed to ionizing radiation as well as in sporadic cases (55,56,57,58,59,60,61). The fusion proteins generated by these recombinant genes homodimerize in a ligand-independent manner through the action of coiled-coil motifs in the N-termini donated by the respective upstream fusion partners (62), resulting in constitutive activation of the tyrosine kinase function of Ret.

FIGURE 70B.1. The RET oncogene is activated by intrachromosomal inversions in papillary thyroid cancers. Reciprocal rearrangements of the tyrosine kinase receptor gene RET with partners in the same arm of chromosome 10 are the most common genetic defects in radiation-induced thyroid cancers, and they are also found in a smaller fraction of sporadic cases. Recombination with H4 (left) gives rise to the RET/PTC1 gene, which consists of a fusion between the promoter and N-terminal coding domains of H4 with the tyrosine kinase domain of RET. Recombination with the promoter and N-terminal coding domains of ELE1 with the same region of RET gives rise to RET/PTC3 (right)
RET/PTC rearrangements are found in 5% to 40% of PTC in the adult population (48). By contrast, RET rearrangements are more common in pediatric PTC, and in cancers from children exposed to ionizing radiation (63,64,65,66,67). There are several lines of evidence pointing to RET/PTC as one of the key first steps in thyroid cancer pathogenesis:
1. Thyroid-specific overexpression of either RET/PTC1 (68,69) or RET/PTC3 (70), in transgenic mice leads to development of tumors with histologic features consistent with PTC.
2. There is a high prevalence of RET/PTC expression in occult or microscopic PTC (71,72).
3. Exposure of cell lines (73) and fetal thyroid explants (74) to ionizing radiation results in expression of RET/ PTC within hours, supporting a direct role for radiation in the illegitimate recombination of RET.
4. The breakpoints in the RET and ELE1/RFG genes resulting in the RET/PTC3 rearrangements of radiation-induced pediatric thyroid cancers from Chernobyl are consistent with direct double-stranded DNA break, resulting in illegitimate reciprocal recombination (75).
Moreover, the H4 and RET genes, although lying at a considerable linear distance from each other within chromosome 10, are spatially juxtaposed during interphase in thyroid cells and presumably present a target for simultaneous double strand breaks in each gene after ionizing radiation, thus giving rise to the RET/PTC1 rearrangement (76). Several groups have examined whether RET/PTC may serve as a prognostic marker for papillary thyroid cancers, and although the results are controversial, the preponderance of the evidence is that sporadic PTCs with this rearrangement have a more favorable natural history (77).
Somatic rearrangements of another member of the tyrosine kinase receptor gene family, the protooncogene NTRK1, which codes for the receptor for nerve growth factor (NGF), are also seen in papillary thyroid cancers, albeit with a far lower prevalence (78). NTRK1 encodes for the receptor for NGF. Several thyroid-specific TRK oncogenes, named TRK, TRK-T1, TRK-T2, and TRK-T3, have been isolated (79,80,81). All contain a variable portion of NTRK1, including the tyrosine kinase domain, and differ in the 5′-upstream sequences, contributed by the tropomyosin (TPM3), translocated promoter region (TPR), and TRK-fused (TFG) genes, respectively. Targeted overexpression of TRK-T1 induces lesions resembling PTC in transgenic mice, supporting a role for this oncogene as an alternative pathway in tumor initiation (82).
The RET gene also plays a prominent role in the pathogenesis of medullary thyroid cancer. Germline mutations of RET confer predisposition to all forms of familial medullary thyroid carcinoma (MTC), either as part of the multiple endocrine neoplasia type 2 (MEN-2) syndromes or when MTC is the sole manifestation of the disease (83,84,85). Acquired somatic mutations of RET are also found in a large fraction of sporadic medullary thyroid carcinomas. RET is activated by point mutations in MTC, resulting in a gain of function of the receptor. The genetic basis of MTC is of great clinical relevance, and genotyping is used as a basis for prophylactic thyroidectomy in RET mutation gene carriers. Readers are referred to Chapter 71 for a more comprehensive discussion of the molecular genetics of RET in MTC, genotype/phenotype correlations, and relevance to patient management.
G Protein–Coupled Receptors
Growth of pituitary, thyroid, and adrenal cells is controlled in part through polypeptide ligands that activate G protein–coupled receptors of the seven-transmembrane domain family, and that signal in part through stimulation of adenylyl cyclase activity. The TSH receptor (TSHR) can be activated by point mutations leading to amino acid substitutions that either abrogate the requirement for ligand or enhance responses to ligand-mediated activation (86). Germline mutations of this receptor are associated with syndromes of familial hyperthyroidism (87). More relevant to neoplasia, somatic mutations of the TSHR are observed in many autonomously functioning thyroid nodules (AFTNs). These benign tumors are characterized by progressive growth and the ability to synthesize hormones in the absence of TSH stimulation. Activating mutations of TSHR result in amino acid substitutions at multiple sites of the receptor protein, including the third, sixth, and seventh transmembrane domains, and the first, second, and third intracellular loops (88). Mutations of TSHR are believed to release the receptor protein from structural constraints present in its unliganded form, thus allowing it to activate Gsα in a constitutive manner (89). Although there is general agreement that TSHR mutations play an important role in pathogenesis of AFTNs, there is some debate about the actual prevalence of these abnormalities (90,91). About 50% to 80% of solitary AFTNs have TSHR mutations, whereas in autonomous nodules present in multinodular goiters the frequency appears to be less (92). The consensus from clinical studies is that AFTNs have a low probability of malignant transformation (93). Accordingly, activating point mutations of TSHR are rare in differentiated thyroid carcinomas (94,95,96,97,98,99,100).
Nuclear Receptors
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that bind to DNA as heterodimers with the retinoid X receptors. The activity of PPARγ is regulated by binding to small lipophilic ligands, mainly fatty acids derived from nutritional intake or metabolism (101). PPARγ has been shown to play an important role in regulating genes involved in adipocyte differentiation and lipid metabolism. An involvement of this nuclear receptor in thyroid cancer was recently discovered by the characterization of a translocation in a subset of human thyroid follicular carcinomas, t(2;3)(q13;p25), which results in fusion of the DNA binding domains of the thyroid transcription factor PAX8 to domains A to F of PPARγ1 (102). PAX8-PPARγ1 messenger RNA (mRNA) and protein are present specifically in follicular neoplasms, particularly in follicular carcinomas. Based on the studies published so far, prevalence of the PAX8-PPARγ1 rearrangement is 10% (11/112) in follicular adenomas, and 41% (34/82) in follicular carcinomas (102,103,104,105,106), suggesting a role for this fusion protein in malignant transformation. The mechanism of transformation induced by PAX8-PPARγ1 is unclear, although it has been shown to have a dominant negative effect on thiazolidinedione-induced transactivation by PPARγ1 (102). Overexpression of wild-type PPARγ1 in thyroid cancer cell lines inhibits cell growth, an effect that is further enhanced by PPARγ1 agonists (107). Taken together, these data suggest that the PAX8- PPARγ1 fusion protein may derepress growth inhibitory controls requiring PPARγ1.
Recently, a remarkably high prevalence (>90%) of mutant of thyroid hormone receptor (TR) α1 and β1 transcripts was reported in human papillary thyroid carcinomas (108). Many of these mutant TRs had impaired transactivation properties, and were predicted to act in a dominant negative fashion. However, these findings were not confirmed in a separate independent study, in which no such mutations were found despite full-length sequencing of TRβ1 complementary DNAs from a similar number of thyroid cancers (109). Homozygous gene–targeted mice with a mutant TR β (TR βPV mice) develop invasive thyroid cancers late in life (110). Although this is consistent with a role for TRβ in thyroid cancer progression, it should be noted that these mice have extreme thyroid hormone resistance and high TSH levels throughout life. In summary, a role for TR loss-of-function mutations in thyroid cancer is intriguing but remains controversial.
DEFECTS IN THE INTRACELLULAR TRANSMISSION OF THE SIGNAL
RAS Oncogenes
After ligand binding, tyrosine kinase receptors dimerize and become autophosphorylated on selected tyrosine residues within their cytoplasmic region. The phosphorylated tyrosines create high-affinity binding sites for proteins containing Src homology 2 (SH2) domains. Recruitment of SH2-containing proteins represents the initial link in a chain of phosphorylation reactions that relay and amplify the growth factor receptor signal. Coupling to Ras takes place through the SH2/SH3 adaptor protein Grb2, which is in turn linked to Sos proteins that activate Ras by exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP). GTP-bound Ras is inactivated by hydrolysis of its associated GTP to GDP, which is dependent on its intrinsic GTPase activity, and on interaction with proteins (GTPase-activating proteins, or GAPs) that markedly increase the hydrolysis of Ras-bound GTP. Three RAS genes are prominent in cancer pathogenesis: H-RAS, K-RAS and N-RAS. From a biochemical standpoint their properties are quite similar, and all can be converted to an oncogenic form. Point mutations in discrete domains within RAS that increase protein affinity for GTP, or inactivate its GTPase function, permanently switch the protein to the “on” position. Activation of Ras leads to the activation of mitogen-activated protein (MAP) kinases, thus linking the activity of cell surface receptors to critical intracellular targets. MAP kinase activity is regulated through the sequential activation of three kinases, which in the case of extracellular signal–related kinases (ERKs) includes successive activation of Raf and MEK kinases. As discussed in greater detail below, this pathway is believed to be central to Ras-mediated dedifferentiation and transformation (111,112). Remarkably, 30% of all human tumors contain a mutation in a RAS allele, making RAS the most widely mutated human protooncogene (113). Mutations of all three RAS oncogenes have been reported in thyroid neoplasms (prevalence is N-RAS > H-RAS > K-RAS) (114,115). RAS mutations are fairly common in both follicular adenomas and carcinomas, but are more prevalent in the latter (116,117,118). In addition, they are present in a small subset of papillary carcinomas (114,119), particularly in follicular variant PTCs (120). The fact that RAS mutations are present in both benign and malignant thyroid neoplasms suggests that RAS activation may be an early step in thyroid tumor development (119,121). In support of this, transgenic mice with thyroid-specific expression of mutant K-Ras develop benign thyroid lesions (122), which tend to become more prevalent and undergo malignant transformation after prolonged goitrogen stimulation. Despite early onset of Ras activation in human thyroid tumor development and progression, RAS mutations may confer cancers with more aggressive properties, because undifferentiated and anaplastic cancers have a higher prevalence of these genetic defects (116,123).
The functional consequences of mutagenic Ras activation in thyrocytes are only partially understood. Activation of Ras induces cell proliferation in primary cultures of human thyrocytes (124,125) and in rat thyroid cell lines (126,127). In human thyroid cells Ras-induced proliferation is self-limited through a mechanism that may require reactivation of expression of the cyclin-dependent kinase inhibitor p16(INK4a) (124,128). By contrast, in immortalized rat thyroid cell lines, Ras-induced cell proliferation is followed by genomic instability (129) and apoptosis (126,127). Mutant Ras is not by itself sufficient to induce thyroid cell transformation, as determined by the ability to form tumors in athymic mice or colonies in soft agar (130). In primary cultures of human thyroid cells, Ras activation does not impair thyroid-differentiated function, whereas in immortalized rat thyroid cell lines, Ras inhibits TSH-induced iodine uptake, and expression of thyroglobulin (Tg) and thyroid peroxidase (TPO) (131). The differences between human thyroid cells and immortalized rat thyroid cell lines suggest that Ras requires an as yet unidentified cooperating event to induce thyroid dedifferentiation. Ras-mediated dedifferentiation in PCCL3 cells requires Raf-MEK-ERK (111,112), and may occur in part by interference with cyclic adenosine monophosphate (cAMP)–dependent signaling, through exclusion of the catalytic domain of protein kinase A from the nucleus (132,133). Ras activation interferes with phosphorylation and activation of TTF-1, a transcription factor necessary for expression of thyroid-specific genes such as the TSH receptor, thyroglobulin, and thyroid peroxidase (111).
BRAF
There are three isoforms of the serine-threonine kinase Raf in mammalian cells: ARaf, BRaf, and CRaf or Raf1, with different tissue distribution of expression (134). Although all Raf isoforms activate MEK phosphorylation, they are differentially activated by oncogenic Ras. In addition, BRaf has higher affinity for MEK1 and MEK2 and is more efficient in phosphorylating MEKs than other RAF isoforms (135). BRAF somatic mutations were recently reported in a high proportion of benign nevi (136) and malignant melanomas (137), and in a smaller subset of colorectal and ovarian cancers (137). A total of 98% of the mutations in melanomas resulted from thymine-to-adenine transversions at nucleotide position 1796, resulting in a valine-to-glutamate substitution at residue 599 (V599E). This mutation is believed to mimic the phosphorylation in the activation segment by insertion of an acidic residue close to a site of regulated phosphorylation at serine 598. BRafV599E exhibits elevated basal kinase activity, and has diminished responsiveness to stimulation by oncogenic H-Ras. BRafV599E also transformed NIH3T3 cells with higher efficiency than the wild-type form of the kinase, consistent with it functioning as an oncogene. This same somatic mutation of BRAF is the most common genetic change in PTC, and present in about 40% of cases (138,139,140,141,142,143,144,145). BRAFT1796Amutations are unique to PTC, and not found in any other form of well-differentiated follicular neoplasm arising from the same cell type. There is practically no overlap between PTC with RET/PTC, BRAF, or RAS mutations, which altogether are found in about 70% of cases (138,144). The lack of concordance for these mutations provides compelling genetic evidence for the requirement of this signaling system for transformation to PTC. Because these signaling proteins function along the same pathway in thyroid cells, this represents a unique paradigm of human tumorigenesis through mutation of three signaling effectors lying in tandem (Fig. 70B.2). BRAF mutations can occur early in development of PTC, based on evidence that they are present in microscopic PTC (140). Moreover, PTC with BRAF mutations have more aggressive properties and can transform to undifferentiated or anaplastic carcinomas (140,142), whereas PTC with RET/ PTC rearrangements do not.

FIGURE 70B.2. Papillary thyroid cancers have non-overlapping mutations of RET/PTC, RAS, or BRAF. Signaling via RAS-RAF-MAP kinase may be required for some of the transforming properties of the RET/PTC oncoproteins in vitro (left). Three of the effectors in this pathway—RET/PTC, RAS, and B-RAF (stars)—can be activated by mutations in papillary carcinomas. Surveys of papillary carcinoma tissue samples showed no overlap in mutation of these genes (right), providing strong genetic evidence for the requirement of this pathway in thyroid tumorigenesis. (Right panel reproduced from Kimura ET, Nikiforova MN, Zhu Z, et al. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 2003;63:1454–1457, with permission.)
The GSP Oncogene
As discussed, TSHR is subject to somatic activating mutations in autonomously functioning thyroid adenomas, indicating that intermediates along the cAMP-dependent signal transduction cascade can function as oncogenes when their structure is modified in a manner that renders them constitutively active. After ligand activation, the TSHR associates with the heterotrimeric G-protein complex, which in turn transmits the signal by stimulating the catalytic activity of adenylyl cyclase. The Gsα subunit of this complex is also a target of activating point mutations in autonomously functioning thyroid adenomas (146,147), as well as in growth hormone–secreting pituitary tumors (148). The resulting GSP oncogene has mutations that are confined to one of two possible hot spots, which either inactivate the intrinsic GTPase activity of the protein (Arg201), or alter the affinity of Gsα for guanine nucleotides (Gln227). Mutations of GSα are uncommon in thyroid carcinomas (97,98,147), again attesting to the fact that constitutive activation of intermediates along the adenylyl cyclase signaling cascade may not be a frequent harbinger of malignant transformation.
β-catenin
β-catenin is a ubiquitously expressed cytoplasmic protein involved in cell-cell adhesion and regulation of gene transcription as a downstream signaling molecule in the wingless pathway. Inappropriate stabilization of β-catenin followed by nuclear translocation has been proposed as an important step in oncogenesis. Stabilization may occur through activating mutations in exon 3 at the phosphorylation sites for ubiquitination and degradation of β-catenin. Somatic mutations in exon 3 of β-catenin associated with nuclear localization were reported in about 25% of poorly differentiated and 60% of anaplastic carcinomas (149,150), but not in well-differentiated cancers (151), strongly implicating the Wnt-β catenin pathway in late-stage progression of thyroid cancer (152).
Other Candidate Oncogenes
The oncogenes discussed so far have been shown to play a role in the pathogenesis of thyroid neoplasms based on the fact that they are subject to genetic modifications that are selected during tumor microevolution. In some cases, there is controversy as to whether a particular oncogene is subject to mutations in thyroid cancer. Other signaling effectors may play critically important roles, and be required for oncogenesis, but not be subject to illegitimate activation through mutations. Because of space limitations these will not be described here, and the reader is referred to other papers and reviews for a more expanded discussion (153,154,155,156,157).
TUMOR SUPPRESSOR GENES
Tumor suppressor genes code for proteins that normally inhibit or restrict cell division. Cells must overcome a series of barriers that normally restrain growth in order to progress toward malignancy. Whereas oncogenes act in a dominant fashion, that is, a disruption in one allele is sufficient to promote the neoplastic phenotype, tumor suppressor genes have a recessive mode of action. In familial syndromes, such as in congenital retinoblastoma or MEN-1, one of the alleles of the tumor suppressor gene (i.e., RB, MENIN) is already mutated in the germline, but cells remain normal unless the second allele is also damaged, which often takes place postnatally. Structural inactivation of both alleles of a tumor suppressor gene can also arise as an acquired event during the course of sporadic tumor evolution.
Retinoblastoma
The normal counterparts of several tumor suppressor genes play important roles in control of cell cycle progression. The retinoblastoma (RB) gene product, the first tumor suppressor to be identified (158), serves as a gatekeeper between the G1 and S phases of the cell cycle. During G1, Rb is underphosphorylated, and prevents entry into S by binding and sequestering transcription factors needed to activate DNA synthesis. Rb is inactivated by phosphorylation when the cell has completed the preparation to replicate its DNA (159). Inactivating mutations of both copies of RB illegitimately release this cell cycle block. RB mutations are seen not only in retinoblastomas, but in common cancers as well. Viral oncoproteins such as the SV40 large T antigen, adenoviral E1A, and the human papilloma virus E7 can also functionally inactivate Rb, in part by their ability to bind to the underphosphorylated domains of the protein (160,161,162). Cancer cell lines lacking functional Rb can be growth arrested by reintroduction of a normal copy of the gene (163). Although mice with homozygous germline inactivation of RB are not viable, heterozygous RB-null mice develop intermediate lobe pituitary tumors (164), and when crossed with P53-null heterozygotes, they also develop medullary thyroid cancers (165). A role for RB mutations in development of human medullary thyroid cancers has not been established firmly, although clonal loss of RB alleles has been reported in early stages of development of familial forms of the disease. In tumors derived from thyroid follicular cells, the role of Rb is controversial. Targeted expression of the E7 oncoprotein of human papilloma virus to thyroid cells of transgenic mice is associated with development of a colloid goiter, and progressive occurrence of differentiated malignant lesions, indirectly implicating Rb inactivation in this process (166). However, in human thyroid cancers RB gene allelic losses are either absent (167) or relatively infrequent (168,169). One study reported RB gene mutations, but it remains unclear whether both alleles were inactivated (170). In addition, a subset of thyroid cancers have decreased immunohistochemical staining for Rb (171), but this again is controversial (172).
p16INK4A
The D-type cyclins, in association with the cyclin-dependent kinases cdk4 and cdk6, promote progression through the G1 phase of the cell cycle. p15INK4b and p16INK4a are cyclin-dependent kinase inhibitors that prevent Rb phosphorylation by specifically antagonizing the activity of the cyclin D/cdk4 or cdk6 holoenzymes. Loss of function of p15INK4b or p16INK4a impairs the G1/S cell cycle checkpoint and contributes to the transformation of several cell types. Mutations of P16 have been found in many thyroid cancer cell lines, but only rarely in primary tumors (173,174,175,176). Interestingly, the P16 gene is frequently hypermethylated in thyroid cancers, suggesting that loss of expression of this critical cell cycle checkpoint gene may occur more commonly as an epigenetic event during tumor evolution (176,177,178).
The p53 Tumor Suppressor Gene
P53 is the most commonly mutated gene in human cancer (179). The fact that many cancers have lost p53 function points to the cardinal role it plays in the maintenance of cell homeostasis. The p53 protein is a transcriptional activator, and this property is needed for preservation of its tumor suppressing properties. Most inactivating mutations of the gene nullify one of the sequence-specific DNA binding domains of the protein. In some cell types, a single mutant P53 allele can generate a product that complexes with the wild-type form of the protein to functionally inactivate it. In this circumstance, p53 has a dominant negative effect, and violates the paradigm that tumor suppressor genes are invariably recessive (i.e., both copies must be mutated for the phenotype to manifest). The p53 protein is involved in control of cell cycle progression, largely by its ability to transactivate expression of genes coding for proteins such as p21/WAF1, that induce G1 arrest by inhibiting cyclin-dependent kinase complexes (180,181). In addition, p53 can help trigger a program of apoptosis. The ability to arrest the cell cycle, and under certain conditions to activate a program of cell death, place p53 at a major crossroads in the determination of cell survival. A fuller picture emerges by understanding the conditions that lead to p53 expression. Levels of p53 increase after exposure to agents that induce DNA damage, such as ionizing radiation and certain drugs used in cancer chemotherapy. Presumably, p53 acts to allow DNA repair to proceed under more favorable conditions (182). However, if the damage is overwhelming, p53 can initiate apoptosis to prevent perpetuation of the flawed cell. Interestingly, mice with homozygous disruption of both P53 alleles develop normally, but get cancers at many sites after birth (183). Affected members of families with Li-Fraumeni syndrome inherit a single mutant P53 allele, and have a high predisposition to cancers of several organs (these tumors exhibit somatic mutations in the other P53 allele). Thus, it appears that p53 is required in cellular emergencies. In its absence, cells that would normally be removed survive, and occasionally give rise to cancers.
Inactivating point mutations of the P53 tumor suppressor gene are highly prevalent in anaplastic and poorly differentiated thyroid tumors, but not in well-differentiated papillary or follicular carcinomas (184,185,186,187). These data implicate p53 inactivation as an important step in late-stage progression of thyroid cancer. Besides impairing apoptosis and removing an important cell cycle checkpoint, loss of function of p53 predisposes cells to additional genetic damage, and is therefore commonly associated with cancers displaying aggressive behavior (188). In addition, loss of function of p53 may impair thyroid cell–differentiated gene expression. Introduction of mutant p53 expression vectors into the well-differentiated thyroid cell line PCCL3 results in loss of expression of thyroglobulin, thyroid peroxidase, and the TSH receptor, and preferential impairment of expression of the thyroid-specific transcription factor Pax-8 (189). Conversely, reexpression of wild-type p53 in undifferentiated thyroid carcinoma cell lines is associated with restoration of Pax-8 production, and of expression of thyroid peroxidase (190,191). The precise mechanism by which loss of function of p53 interferes with thyroid-specific gene expression is not known. However, because p53 does not appear to be required for thyroid development, it is likely that loss of function of p53 does not directly impact thyroid-differentiated gene expression.
PTEN
Germline mutations of the PTEN gene confer predisposition to Cowden's disease, a condition characterized by development of hamartomas in multiple organs, and increased risk of breast, thyroid, and other cancers (192,193). PTEN codes for a dual specificity lipid and tyrosine phosphatase, which dephosphorylates phosphatidylinositol 3,4,5 trisphosphate and antagonizes the phosphatidylinositol-3 kinase (PI3K)-protein kinase B (Akt) signaling pathway. Hamartomas and cancers developing in these patients acquire loss-of-function mutations or deletions affecting the remaining PTEN allele (194,195). A role for PTEN on development of sporadic thyroid neoplasms has not yet been firmly established. Allelic loss of the PTEN locus in 10q23.3 is seen in about a quarter of sporadic nonmedullary thyroid cancers, yet this is not coupled with mutations in the hemizygous PTEN allele. Loss or reduction of PTEN protein expression as well as inappropriate subcellular compartmentalization has been reported in nonmedullary thyroid cancers (196,197,198), and may occur in part through epigenetic silencing. The absence of somatic PTEN mutations in thyroid cancer has prompted a screen for other candidate tumor suppressor genes close to the PTEN locus on 10q23.3. MINPP1 has also been localized to this region, and codes for a phosphatase that has the ability to remove the 3-phosphate from inositol phosphate substrates, a function that overlaps that of PTEN. One follicular carcinoma with loss of heterozygosity at this locus had a somatic point mutation in the other MINPP1 allele, but the functional consequences of this are not known (199). Despite these issues, the potential relevance of the PI3K-Akt pathway in thyroid tumorigenesis is supported by the observation that Akt expression and activation is increased in thyroid cancers, particularly in follicular carcinomas (200).
Other Candidate Tumor Suppressor Genes
As mentioned above, loss of function of tumor suppressor genes usually requires structural inactivation of both alleles. One of these is usually lost as part of a large deletion of chromosomal material. Based on this paradigm, the hunt for tumor suppressor genes has been conducted using approaches to identify regions of chromosomal loss that are common in particular forms of tumors. Papillary carcinomas have a low prevalence of chromosomal or allelic losses (201,202). By contrast, follicular neoplasms, particularly follicular carcinomas, exhibit more frequent allelic deletions (202,203). Using polymorphic molecular probes to scan the genome, some of the regions preferentially affected lie within chromosomes 2p, 2q, 3p, 7q, 10q, 11q, and 17p (202,203,204,205,206,207,208). There has been considerable effort to identify potential tumor suppressor genes lying within those regions. No mutations of either the von Hippel-Lindau (VHL) or the FHIT genes were found in follicular neoplasms with 3p deletions, possibly excluding these candidate tumor suppressors as significant in the pathogenesis of differentiated thyroid cancer (204).
Epigenetic Changes: Abnormalities in DNA Methylation
The majority of human gene promoters lie within chromosome areas rich in CG dinucleotides, referred to as CG islands. Most of these CG sites are demethylated, with the exception of genes located on the X chromosome, and certain imprinted loci (209). CG island methylation results in heritable inhibition of gene transcription and has been proposed as an alternative mechanism of gene inactivation in neoplasia (210,211). Increased DNA methylation has been reported to inhibit expression of tumor suppressor genes (212) and to predispose to transitional point mutations at methylated cytosines (213). Abnormal CpG methylation is a common event in neoplasia, usually occurring as an early event (211,214). The mechanism for these methylation changes is unknown. Thyroid tumors are no exception to this paradigm, because they exhibit a high prevalence of methylation abnormalities, occurring early in tumor progression and affecting loci of potential pathogenetic significance (176,177,178,215,216,217,218).
DISRUPTION OF PROGRAMMED CELL DEATH
Tissue homeostasis is maintained in part by factors that control the appropriate balance between cell proliferation and cell death. Apoptosis is a physiologic process that requires sequential expression of gene products that induce chromatin condensation, DNA cleavage, and cytosolic shrinkage. Within endocrine tissues, for instance, apoptosis is required for breast and prostatic involution after lactation, and androgen withdrawal, respectively. As mentioned previously, apoptosis is triggered to prevent proliferation of damaged cells, such as occurs after exposure to ionizing radiation. If tumors are to arise they must successfully bypass the cell-killing program that is activated in response to particular cues. In certain cancers, mutations of genes involved in the regulation of cell death appear to be primary mediators of tumorigenesis. Perhaps the most significant example is the overexpression of bcl-2 in follicular lymphomas that occurs as a result of a translocation between the immunoglobulin heavy chain gene promoter and the bcl-2 gene (219,220). The normal function of bcl-2 in B-lymphocytes is to promote cell survival, presumably as a mechanism to perpetuate immune memory. Illegitimate overexpression of bcl-2 is not in itself sufficient to induce tumorigenesis, but favors the perpetuation of a neoplastic clone by impairing apoptosis, and allowing the survival of cells that accumulate mutations affecting genes involved in growth control. There is presently only sketchy information on the possible role of apoptosis in thyroid tumor evolution. However, initiation of programmed cell death is a key protective mechanism evoked after oncogene activation in thyroid cells (125,126,221,222). For tumor clones to develop and expand, it is likely that the apoptotic program must be disabled through secondary genetic or epigenetic events that are yet to be identified.
FACTORS THAT MAINTAIN THE INTEGRITY OF THE GENOME
Mismatch Repair Deficiency Causing Microsatellite Instability
Hereditary nonpolyposis colorectal cancer (HNPCC) is an autosomal-dominant disease associated with early onset of colorectal cancer. The genes responsible for conferring this predisposition, hMSH2, hMLH1, hPMS1, and hPMS2, code for enzymes that correct nucleotide mismatches that occur during DNA synthesis (223,224). A homozygous disruption of one of these genes (one allele mutated in the germline, the other as an acquired somatic event) results in microsatellite instability (RER), a tendency to acquire alterations at mononucleotide, dinucleotide, or trinucleotide repeat sequences (microsatellites), which are widespread throughout the genome. These regions are more prone to error during DNA replication, and the mismatches cannot be efficiently corrected when the appropriate enzymatic complex is defective. It is thought that the RER genome results in mutations of genes important in growth control, as demonstrated for the TGF βII receptor in colorectal cancers (225). Microsatellite instability is uncommon in sporadic papillary thyroid carcinomas (226,227,228,229,230,231), and probably not a major mechanism of transformation in follicular neoplasms, although the latter findings are not conclusive (227). Whether radiation-induced papillary thyroid cancers exhibit microsatellite instability is somewhat controversial, with some studies failing to find evidence of RER (226), and others finding a low prevalence (231). Perhaps tellingly, patients with HNPCC do not appear to have a higher incidence of thyroid cancer, and none of the studies of sporadic or radiation-induced thyroid cancers has identified mutations of any of the genes coding for mismatch repair enzymes.
Chromosomal Instability
Genetic instability in cancer more commonly manifests as alterations in chromosome number (aneuploidy) (232,233). Follicular carcinomas are more frequently aneuploid than papillary carcinomas, a phenomenon that has been proposed to be due to chromosome nondisjunction (201,234,235). There is reason to believe that aneuploidy may have a genetic etiology, based primarily on studies in yeast in which mutations of over 100 genes can cause chromosomal instability. These include genes involved in chromosome metabolism, spindle assembly, chromosome segregation, and checkpoint control (236). In human cancers, this notion has gained experimental support (232). A predisposition to gain or lose whole chromosomes in colorectal cell lines has been linked to abnormalities in the mitotic checkpoint (237), which is activated by the presence of unattached kinetochores (238). In Saccharomyces cerevisiae, the “budding uninhibited by benzimidazole” (BUB1, 2, and 3), and the mitotic-arrest defective (MAD1, 2, and 3) proteins are intermediates in signal transduction cascades that are activated by mitotic spindle damage and delay onset of anaphase (239,240). This pathway also responds to defects in the structure of centromeres and kinetochores, and is thought to be responsible for the mitotic checkpoint activated by unattached or misaligned chromosomes. In Drosophila, animals with near-null mutations of BUB1 die during late larval stages due to mitotic abnormalities, indicative of bypass of the mitotic checkpoint function (241). Mutations of a human homologue of BUB1 have been identified in a subset of colorectal cancer cell lines with chromosomal instability (237), and in one sporadic rectal cancer (242). Somatic mutations of hBUB1 have also been reported in primary lung cancers and lung cancer cell lines (243). However, subsequent studies have not found this to be a common abnormality in these and other tumor types (244,245). This is also the case in aneuploid thyroid neoplasms and thyroid cancer cell lines with mitotic checkpoint dysfunction, in which mutations of BUB1 or its homologue BUBR1 are largely absent (246). It remains to be seen whether or not somatic mutations of other genes coding for components of the mitotic checkpoint pathway may play a role in aneuploidy. An alternative explanation for the chromosomal instability of colorectal cancers has recently been advanced based on the discovery of a new property of the adenomatous polyposis coli tumor suppressor gene product (APC). A complex of proteins that includes APC may function as adaptors that allow the kinetochore to attach to microtubules. Mutations in APC domains involved in these associations may disrupt normal chromosome alignment and segregation in metaphase, thus predisposing to polyploidy (247).
Overexpression of the pituitary tumor transforming gene product (PTTG), the mammalian homologue of the yeast protein securin, has been reported in different tumor types, including pituitary adenomas (248,249) and thyroid neoplasms (250). Separation of sister chromatids in anaphase is mediated by separase, an endopeptidase that cleaves the chromosomal cohesin complexes that bridge the aligned sister chromatids. Separase is in turn inhibited by securin, which is degraded at the metaphase–anaphase transition. Overexpression of PTTG/securin induces aneuploidy in certain cell types (251). Curiously, mice lacking PTTG/securin are viable, and embryonic PTGG (-/-) fibroblasts have numerous chromosomal abnormalities (252). Thus, disruption of factors involved in sister chromatid separation can result in aneuploidy. So far there are no reports of tumor-promoting mutations of PTTG/securin, and most cancers exhibit primarily higher levels of expression. Interestingly, PTTG/securin overexpression in cell lines induces FGF2 production, and increased abundance of these gene products is a negative prognostic indicator in differentiated thyroid cancers (253).
Some of the early genetic events involved in tumor initiation could confer cells with a “mutator” phenotype, characterized by loss of genomic stability and a higher rate of mutations than normal cells (254). The standard interpretation of this hypothesis is that genes conferring cells with a “mutator” phenotype are likely to be directly involved in DNA repair (e.g., mismatch repair enzymes) or in cell cycle checkpoints. However, there is evidence that constitutive activation of oncogenes such as Ras may also disrupt genomic stability. A key role for Ras in mitotic spindle assembly has been reported in fission yeast (255,256). In mammalian cells, MAP kinases may also play a direct role in mitosis and chromosome segregation. Activated MAP kinase localizes to kinetochores in early and midmitosis, in asters during all stages of mitosis, and in the chromosome midbody in late anaphase (257). Evidence for a temporal sequence of localization of activated MAP kinase in different nuclear compartments during mitosis (257,258) suggests that phosphorylation-dephosphorylation steps are needed for orderly progression, a step that may be disrupted when MAPK activation is constitutive. Induction of expression of the human H-Ras oncogene in p53-null cells leads to premature entry of cells into S phase, increased permissivity for gene amplification, and generation of aberrant chromosomes within a single cell cycle (259,260). Overexpression of oncogenic Ras has also been shown to produce chromosome aberrations in rat mammary carcinoma cells (261), rat prostatic tumor cells (262), and in a human colon carcinoma cell line (263). In thyroid PCCL3 cells, acute H-Rasv12 activation induces chromosome missegregation, centrosome amplification, and chromosome misalignment within two cell cycles after activation (129). A putative role of RAS as a mutator gene has not been studied in animal models or in human studies.
EVENTS PREDISPOSING TO THYROID NEOPLASIA
Genetic Predisposition to Nonmedullary Thyroid Cancer
There are several familial syndromes of neoplasia that exhibit increased incidence of tumors of thyroid follicular cells (Table 70B.1). An increased predisposition to papillary carcinoma is well established in certain families with adenomatous polyposis (FAP), an autosomal condition characterized by the presence of multiple adenomatous polyps of the intestine (264,265,266). The association with thyroid cancer extends to Gardner's syndrome, a variant of FAP characterized by numerous intestinal adenomas, osteomas, soft tissue lesions, and other extracolonic neoplasms (267,268). Thyroid cancers in FAP exhibit a marked female preponderance (female:male ratio 8:1) and are more common under the age of 30. Women under 35 years of age with FAP have been estimated to have a 160-fold higher risk for thyroid carcinoma than normal individuals (265). Papillary carcinomas from patients with FAP are commonly multifocal, well encapsulated, and often display unusual histopathologic features, such as areas of cribriform, solid, and spindle cells within the tumors (269). Predisposition to FAP is conferred by germline-inactivating mutations of APC, which maps to chromosome 5q21 (270,271). Colorectal neoplasms from patients with FAP frequently exhibit loss of heterozygosity at this locus, consistent with a role for APC as a tumor suppressor gene, requiring loss of function of both alleles in order for the recessive phenotype to emerge. However, no biallelic inactivation of the APC gene was found in thyroid cancers from four patients in two FAP kindreds (272). Most patients with FAP developing papillary thyroid carcinomas have APC defects lying outside of the mutation cluster region (codons 1286–1513), considered the hot spot mutation area for extracolonic manifestations of FAP (273). The existence of “modifier” genes that act in concert with APC to alter the predisposition to tumor formation at extracolonic sites, such as the thyroid gland, has been proposed. This is supported by analysis of FAP kindreds with family members inheriting the same APC mutation but differing dramatically in tumor burden (i.e., number of polyps). It is noteworthy that mutations of APC are not common in sporadic thyroid cancers, suggesting that inactivation of this gene is not likely to be a major pathogenetic event in thyroid tumorigenesis (274,275).
TABLE 70B.1. FAMILIAL SYNDROMES ASSOCIATED WITH TUMORS OF THYROID FOLLICULAR CELLS
Disease Entity
Manifestations
Chromosomal Location
Gene
Function
Familial polyposis coli
Polyps in large intestine, papillary thyroid cancer
5q21
APC
Cell adhesion, mitotic checkpoint
Gardner's syndrome
Polyps in small and large intestine, osteomas, fibromas, lipomas, ampullary cancers, papillary thyroid cancer
5q21, others (?)
APC
Cell adhesion, mitotic checkpoint
Turcot's syndrome
Polyps in large intestine, brain tumors, papillary thyroid cancer
5q21, others (?)
APC
Cell adhesion, mitotic checkpoint
Multiple endocrine neoplasia type I
Parathyroid adenomas, pituitary adenomas, pancreatic endocrine tumors, follicular adenomas (?)
11q13
Menin
Transcriptional regulation, telomerase activity
Cowden's disease
Multiple hamartomas, follicular adenomas, goiter, follicular carcinomas
10q22-23
PTEN
Dual-specificity phosphatase
Carney complex
Spotty skin pigmentation, myxomas, schwannomas, pigmented adrenocortical nodules, hypercortisolism, follicular adenomas, follicular carcinomas
2p16, 17q23
PKARIA
cAMP signaling
Familial non-medullary thyroid cancer syndromes
Papillary thyroid carcinoma, non-toxic multinodular goiter
14q
MNGI (unknown)
Papillary thyroid cancer and papillary renal neoplasms
1q21
PTC (unknown)
Benign and malignant tumors with oxyphilia
19p13.2
TCO (unknown)
Thyroid tumors have also been reported in other familial syndromes. They are a frequent extracutaneous manifestation of Cowden's disease (multiple hamartoma syndrome), being observed in two thirds of patients; they include benign thyroid lesions (adenomas, goiter, thyroglossal duct cyst), and follicular thyroid carcinoma (276). As mentioned above, the gene for Cowden's disease has recently been identified (PTEN), and found to function as a dual-specificity phosphatase. There are case reports describing the association of thyroid carcinoma in patients with Peutz-Jeghers syndrome (277) and ataxia-telangiectasia (278).
It is controversial whether there is a significant increase in prevalence of thyroid tumors in patients with multiple endocrine neoplasia type 1 (MEN-1). When present, thyroid disease is observed mostly as benign lesions (nodular hyperplasia, goiter, adenoma), and far more rarely as a malignancy (279). The gene conferring predisposition to MEN-1 (MENIN) is located on chromosome 11q13 (280,281), and believed to function as a tumor suppressor through mechanisms that may involve interference with control of gene transcription (282,283), DNA replication/repair (284,285), and telomerase activity (286,287). Pancreatic, pituitary, and parathyroid tumors from patients with MEN-1 frequently exhibit loss of heterozygosity at this locus, leading to loss of function of the normal allele. Loss of heterozygosity at chromosome 11q13 is also found in sporadic follicular thyroid neoplasms, but to our knowledge no intragenic MENIN mutations have been reported in these tumors (206,208). However, a role for MENIN in thyroid tumor development is supported by data from heterozygous MENIN knockout mice, which develop thyroid neoplasms (288).
Carney complex is an autosomal-dominant disorder associated with cardiac myxomas and endocrine tumors caused in part by inactivating mutations of the gene encoding the regulatory subunit 1A of the cAMP-dependent protein kinase (PKAR1A), resulting in its inappropriate activation (289,290). Although the Carney complex is primarily associated with benign endocrine neoplasms, thyroid follicular carcinomas may also be observed (291).
There have been multiple reports of familial clustering of papillary carcinomas (292,293,294,295,296). Most of the literature consists of descriptions of individual family pedigrees. This has resulted in uncertainty as to whether the familial association represents evidence of genetic predisposition to the disease, exposure to a common environmental triggering event, increased susceptibility to environmental effects, or a chance occurrence. A comprehensive analysis of families of all cases of papillary carcinoma diagnosed in Iceland between 1955 and 1984 revealed that 3.8% of the propositi had a first-degree relative with thyroid carcinoma, a higher than expected frequency that was not, however, statistically significant (although there was a significantly increased risk in male relatives) (297). Stoffer et al studied families from 226 consecutive papillary thyroid carcinoma patients from a private practice, and concluded that 3.5% to 6.2% had at least one affected relative (298). Ron et al reported a population-based case control study of the Connecticut Tumor Registry, in which a fivefold excess risk for nonmedullary thyroid cancer was found in close relatives (299,300). On balance, the evidence for a genetic basis for familial nonmedullary thyroid carcinoma is quite supportive. The transmission of susceptibility to familial nonmedullary thyroid cancer is compatible with autosomal-dominant inheritance with reduced penetrance, or with complex inheritance (301). Linkage studies have identified the putative locations of three genes for familial NMTC: MNG1 (302), TCO (303,304), and PTC (305). A large Canadian family of German origin showed linkage of MNG1 to chromosome 14q (302). This family included 18 cases of nontoxic multinodular goiter, with 2 individuals having papillary lesions suggestive of papillary thyroid cancer. A gene conferring predisposition to benign and malignant tumors with oxyphilia in a French family was mapped to chromosome 19p13.2 by linkage analysis (303). In a three-generation family with five cases of papillary thyroid cancer, three cases of thyroid nodules, and two cases of papillary renal neoplasia, linkage was found to 1q21 (putative PTC gene) (305).
Radiation-Induced Thyroid Cancer
The major known risk factor for papillary thyroid carcinoma is prior exposure to radiation. The most significant sources of exposure have been after therapeutic irradiation and through environmental disasters (306,307,308). The effects of radiation on thyroid cancer risk are dose dependent (307,308). A lower age at the time of exposure has been consistently associated with a higher relative risk for thyroid cancer, a phenomenon that was clearly apparent in the pediatric thyroid carcinomas arising after the Chernobyl nuclear disaster (309,310,311). As a result of the accident at the Chernobyl nuclear power plant, 8 × 1018 Bq of radioactivity were released, most in the form of radioiodine isotopes. The absorption of radioiodines from ingestion of contaminated food and water and through inhalation led to internal exposure to the thyroid gland, which was 3 to 10 times higher in children than in adults. An increased incidence of thyroid cancer in children from Belarus and Ukraine was noted as early as 4 to 5 years after the accident (312), and the risk was higher in the most contaminated regions. Between 1990 and 1998, the incidence of childhood thyroid cancer in Belarus was at least 60-fold greater than prior to the disaster (309), and this increase extends to contaminated regions of Ukraine and Southwest Russia. More than 95% of post-Chernobyl thyroid cancers are papillary carcinomas. Radiation is known to induce DNA strand breaks, and it was therefore fitting that RET/PTC rearrangements were highly prevalent in cancers from children exposed to radiation after the Chernobyl nuclear accident (64,65,313), or to external irradiation for treatment of benign diseases of the head and neck (64,314,315). It should be noted that RET rearrangements are also found with high prevalence in pediatric papillary thyroid carcinomas with no documented history of radiation exposure, so it cannot be assumed that this genetic recombination is specific to radiation-induced DNA damage (63). There are several lines of evidence indicating that RET/PTC rearrangements are likely caused directly by ionizing irradiation:
1. Irradiation of human fetal thyroid explants, undifferentiated thyroid carcinoma, or fibrosarcoma cells in vitro induces RET/PTC1 rearrangements in a dose-dependent fashion within a short time frame (73,74).
2. The pattern of breakpoints in ELE1 and RET intronic DNA, the target genes involved in the RET/PTC3 recombination, revealed that in each tumor the position of the break in one gene corresponded to the position of the break in the other gene. This tendency suggests that the two genes may lie next to each other in the nucleus. Such a structure would facilitate formation of RET/ PTC3 rearrangements because a single radiation track could produce concerted breaks in both genes, leading to intrachromosomal inversion due to reciprocal exchange via end-joining.
3. Juxtaposition of chromosomal loci participating in the RET/PTC1 rearrangement (i.e., RET and H4) during interphase in normal human thyroid cells was later demonstrated, further suggesting that chromosomal architecture during interphase may be an important prerequisite for RET recombination after radiation in thyroid cells (76) (Fig. 70B.3).
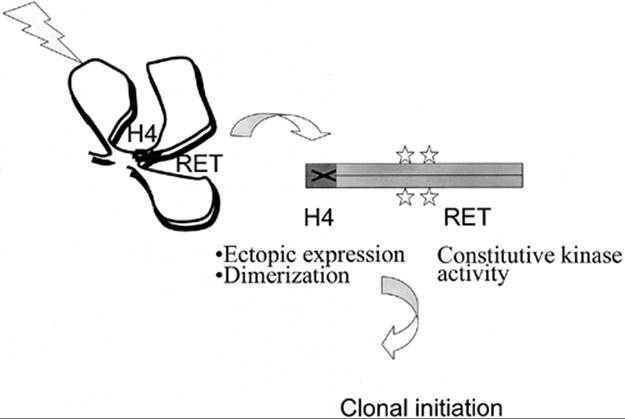
FIGURE 70B.3. Chromosomal architecture during interphase may predispose thyroid cells to RET activation by recombination after radiation exposure. The RET and H4 genes, despite being at a considerable distance from each other on chromosome 10, are spatially juxtaposed during interphase in normal human thyroid cells. This could presumably generate a target for a single radiation track to produce concerted breaks in both genes, leading to intrachromosomal inversion and activation of expression of the H4-RET fusion protein (RET/PTC1). The N-terminal domain of H4 donates a coiled-coil motif, thus allowing constitutive dimerization, and activation of tyrosine kinase activity in an unregulated fashion.
A distinguishing feature of these pediatric radiation-induced tumors is the high prevalence of solid pattern growth manifesting as sheets of malignant epithelial cells surrounded by varying amounts of fibrotic stroma. A solid growth pattern was seen in almost half of all Chernobyl cancers (64). By contrast, this type of histologic appearance is rare in sporadic adult and pediatric papillary carcinomas. Solid variant papillary carcinomas are primarily associated with RET/PTC3, whereas tumors with typical papillary morphology more commonly have RET/PTC1 rearrangements (64,316,317). These data suggest that these two fusion genes may have distinct functional properties. This is supported by the observation that transgenic mice with thyroid-specific expression of RET/PTC1 develop typical papillary carcinomas (68,69), whereas those expressing RET/PTC3 develop solid-variant tumors (318). There are preliminary indications that solid-variant tumors may carry a worse prognosis (319).
Mutations of RAS or P53 were not involved in the generation of radiation-induced PTC (320,321).
CONCLUSIONS: AN INTEGRATED PERSPECTIVE OF THYROID TUMOR PATHOGENESIS
It may be enlightening to attempt to recreate the natural history of thyroid cancers based on available epidemiologic and genetic information. However, it is important to emphasize that there is much we do not know. Progression of thyroid tumors is mostly interrupted when they are removed at surgery, making data on prognostic implications of particular genotypes difficult to evaluate. Although in part speculative, this remains a worthwhile exercise, because it may point to some unresolved questions and areas of opportunity for the future.
Papillary Thyroid Carcinomas
A general assumption in tumor genetics is that a single oncogenic mutation is not sufficient to induce malignant transformation. It follows that sequential abnormalities must take place. The evidence that activating rearrangements of tyrosine kinase receptors (i.e., RET/PTCs and NTRK) are likely early events in papillary thyroid cancer has been reviewed, and is now quite compelling. Similarly, recent studies indicating that papillary cancers harbor mutually exclusive mutations of RET/PTC or RAS or B-RAF point to a requirement for constitutive activation of this signaling pathway (presumably acting via MEK-ERK) in tumor initiation. PTCs harboring different RET/PTC variants, RAS, or BRAF mutations often exhibit distinct pathologic features, and may also differ in their biologic behavior, indicating that the nature of the tumor-initiating event is important. Thus, most solid-variant PTCs harbor the RET/PTC3 rearrangement, follicular variant PTCs commonly have RAS mutations, and tall cell variant PTCs have BRAF mutations. Clearly, other cooperating genetic events must take place, but these are yet to be discovered. There is now an abundance of information on global gene expression changes and epigenetic modifications of putative tumor suppressor genes in PTC, but their significance to tumor progression has not been studied. Papillary carcinomas have a low rate of aneuploidy, loss of heterozygosity, and microsatellite instability. This predicts that the tumor clone will remain fairly homogeneous, and explains in part the excellent prognosis and response to therapy of this type of thyroid cancer.
Follicular Thyroid Carcinomas
Follicular carcinomas are believed to arise from benign adenomas. Absolute proof of the adenoma–carcinoma transition is lacking, but the existence of intermediate phenotypes (i.e., minimally invasive follicular carcinomas) is consistent with this microevolution. Follicular carcinomas are more common in iodine deficiency regions, suggesting that the proliferative drive may be a factor in their development. Mutations of all three mammalian RAS genes (H-RAS, N-RAS, and to a lesser extent K-RAS) are seen in follicular adenomas and carcinomas, although they appear to be more prevalent in the latter. It is unclear why RASmutations are associated with both PTC and FTC. One can speculate that the impact of RAS mutations on tumor phenotype may differ according to whether they occur early or later in tumor progression, and with which other cooperating genetic event they are associated. PAX8-PPARγ1 rearrangements are present specifically in follicular neoplasms, particularly follicular carcinomas. Based on the observation that FTC either have RAS mutations or PAX8-PPAR γ1 rearrangements, but rarely both, the existence of two distinct and virtually nonoverlapping molecular pathways initiated by these oncoproteins has been proposed (322). FTC have higher rates of allelic loss and aneuploidy, consistent with chromosomal instability.
Undifferentiated or Anaplastic Thyroid Carcinomas
The end-stage forms of thyroid cancer are characterized by loss of differentiated properties, greater tumor invasiveness, and metastatic spread. Both FTC and PTC can progress to undifferentiated or anaplastic carcinomas. Mutations of the P53 tumor suppressor gene are a common feature of undifferentiated thyroid cancers, and this tumor suppressor is a strong candidate for the transition from well-differentiated to anaplastic carcinoma.
REFERENCES
1. Namba H, Matsuo K, Fagin JA. Clonal composition of benign and malignant human thyroid tumors. J Clin Invest 1990; 86:120–125.
2. Fey MF, Peter HJ, Hinds HL, et al. Clonal analysis of human tumors with M27 beta, a highly informative polymorphic X chromosomal probe. J Clin Invest 1992;89:1438–1444.
3. Aeschimann S, Kopp PA, Kimura ET, et al. Morphological and functional polymorphism within clonal thyroid nodules. J Clin Endocrinol Metab 1993;77:846–851.
4. Wynford-Thomas D, Stringer BM, Williams ED. Dissociation of growth and function in the rat thyroid during prolonged goitrogen administration. Acta Endocrinol (Copenh) 1982;101: 210–216.
5. Black EG, Logan A, Davis JR, et al. Basic fibroblast growth factor affects DNA synthesis and cell function and activates multiple signalling pathways in rat thyroid FRTL-5 and pituitary GH3 cells. J Endocrinol 1990;127:39–46.
6. Eggo MC, Hopkins JM, Franklyn JA, et al. Expression of fibroblast growth factors in thyroid cancer. J Clin Endocrinol Metab 1995;80:1006–1011.
7. Shingu K, Sugenoya A, Itoh N, et al. Expression of basic fibroblast growth factor in thyroid disorders. World J Surg 1994; 18:500–505.
8. Boelaert K, McCabe CJ, Tannahill LA, et al. Pituitary tumor transforming gene and fibroblast growth factor-2 expression: potential prognostic indicators in differentiated thyroid cancer. J Clin Endocrinol Metab 2003;88:2341–2347.
9. Clement S, Refetoff S, Robaye B, et al. Low TSH requirement and goiter in transgenic mice overexpressing IGF-I and IGF-Ir receptor in the thyroid gland. Endocrinology 2001;142:5131–5139.
10. Vella V, Sciacca L, Pandini G, et al. The IGF system in thyroid cancer: new concepts. Mol Pathol 2001;54:121–124.
11. van der Laan BF, Freeman JL, Asa SL. Expression of growth factors and growth factor receptors in normal and tumorous human thyroid tissues. Thyroid 1995;5:67–73.
12. Belfiore A, Pandini G, Vella V, et al. Insulin/IGF-I hybrid receptors play a major role in IGF-I signaling in thyroid cancer. Biochimie 1999;81:403–407.
13. Lemoine NR, Hughes CM, Gullick WJ, et al. Abnormalities of the EGF receptor system in human thyroid neoplasia. Int J Cancer 1991;49:558–561.
14. Aasland R, Akslen LA, Varhaug JE, et al. Co-expression of the genes encoding transforming growth factor-alpha and its receptor in papillary carcinomas of the thyroid. Int J Cancer 1990; 46:382–387.
15. Colletta G, Cirafici AM, Di Carlo A, et al. Constitutive expression of transforming growth factor alpha does not transform rat thyroid epithelial cells. Oncogene 1991;6:583–587.
16. Dawson TP, Radulescu A, Wynford-Thomas D. Expression of mutant p21ras induces insulin-like growth factor 1 secretion in thyroid epithelial cells. Cancer Res 1995;55:915–920.
17. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2002;2:795–803.
18. Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2002;2:727–739.
19. Jussila L, Alitalo K. Vascular growth factors and lymphangiogenesis. Physiol Rev 2002;82:673–700.
20. Viglietto G, Maglione D, Rambaldi M, et al. Upregulation of vascular endothelial growth factor (VEGF) and downregulation of placenta growth factor (PlGF) associated with malignancy in human thyroid tumors and cell lines. Oncogene 1995; 11:1569–1579.
21. Soh EY, Duh QY, Sobhi SA, et al. Vascular endothelial growth factor expression is higher in differentiated thyroid cancer than in normal or benign thyroid. J Clin Endocrinol Metab 1997;82: 3741–3747.
22. Shushanov S, Bronstein M, Adelaide J, et al. VEGFc and VEGFR3 expression in human thyroid pathologies. Int J Cancer 2000;86:47–52.
23. Hung CJ, Ginzinger DG, Zarnegar R, et al. Expression of vascular endothelial growth factor-C in benign and malignant thyroid tumors. J Clin Endocrinol Metab 2003;88:3694–3699.
24. Friesel RE, Maciag T. Molecular mechanisms of angiogenesis: fibroblast growth factor signal transduction. FASEB J 1995; 9:919–925.
25. Franzen A, Piek E, Westermark B, et al. Expression of transforming growth factor-beta1, activin A, and their receptors in thyroid follicle cells: negative regulation of thyrocyte growth and function. Endocrinology 1999;140:4300–4310.
26. Grubeck-Loebenstein B, Buchan G, Sadeghi R, et al. Transforming growth factor beta regulates thyroid growth. Role in the pathogenesis of nontoxic goiter. J Clin Invest 1989;83:764–770.
27. Kimura ET, Kopp P, Zbaeren J, et al. Expression of transforming growth factor beta1, beta2, and beta3 in multinodular goiters and differentiated thyroid carcinomas: a comparative study. Thyroid 1999;9:119–125.
28. Bhowmick NA, Chytil A, Plieth D, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004;303:848–851.
29. Nose F, Ichikawa T, Fujiwara M, et al. Up-regulation of cyclooxygenase-2 expression in lymphocytic thyroiditis and thyroid tumors: significant correlation with inducible nitric oxide synthase. Am J Clin Pathol 2002;117:546–551.
30. Cornetta AJ, Russell JP, Cunnane M, et al. Cyclooxygenase-2 expression in human thyroid carcinoma and Hashimoto's thyroiditis. Laryngoscope 2002;112:238–242.
31. Specht MC, Tucker ON, Hocever M, et al. Cyclooxygenase-2 expression in thyroid nodules. J Clin Endocrinol Metab 2002; 87:358–363.
32. Bauer AJ, Patel A, Terrell R, et al. Systemic administration of vascular endothelial growth factor monoclonal antibody reduces the growth of papillary thyroid carcinoma in a nude mouse model. Ann Clin Lab Sci 2003;33:192–199.
33. Seno H, Oshima M, Ishikawa TO, et al. Cyclooxygenase 2- and prostaglandin E(2) receptor EP(2)-dependent angiogenesis in Apc(Delta716) mouse intestinal polyps. Cancer Res 2002;62: 506–511.
34. Oshima M, Murai N, Kargman S, et al. Chemoprevention of intestinal polyposis in the Apcdelta716 mouse by rofecoxib, a specific cyclooxygenase-2 inhibitor. Cancer Res 2001;61:1733–1740.
35. Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med 2003;348: 891–899.
36. Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med 2003;348:883–890.
37. Puxeddu E, Mitsutake N, Knauf JA, et al. Microsomal prostaglandin E2 synthase-1 is induced by conditional expression of RET/PTC in thyroid PCCL3 cells through the activation of the MEK-ERK pathway. J Biol Chem 2003;278:52131–52138.
38. Collins VP. Epidermal growth factor receptor gene and its transcripts in glioblastomas. Rec Results Cancer Res 1994;135:17–24.
39. Di Fiore PP, Kraus MH. Mechanisms involving an expanding erbB/EGF receptor family of tyrosine kinases in human neoplasia. Cancer Treat Res 1992;61:139–160.
40. Pegram MD, Konecny G, Slamon DJ. The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat Res 2000;103:57–75.
41. Habib AA, Chun SJ, Neel BG, et al. Increased expression of epidermal growth factor receptor induces sequestration of extracellular signal-related kinases and selective attenuation of specific epidermal growth factor-mediated signal transduction pathways. Mol Cancer Res 2003;1:219–233.
42. Chen BK, Ohtsuki Y, Furihata M, et al. Co-overexpression of p53 protein and epidermal growth factor receptor in human papillary thyroid carcinomas correlated with lymph node metastasis, tumor size and clinicopathologic stage. Int J Oncol 1999;15:893–898.
43. Haugen DR, Akslen LA, Varhaug JE, et al. Expression of c-erbB-3 and c-erbB-4 proteins in papillary thyroid carcinomas. Cancer Res 1996;56:1184–1188.
44. Di Renzo MF, Olivero M, Ferro S, et al. Overexpression of the c-MET/HGF receptor gene in human thyroid carcinomas. Oncogene 1992;7:2549–2553.
45. Belfiore A, Gangemi P, Costantino A, et al. Negative/low expression of the Met/hepatocyte growth factor receptor identifies papillary thyroid carcinomas with high risk of distant metastases. J Clin Endocrinol Metab 1997;82:2322–2328.
46. Ivan M, Bond JA, Prat M, et al. Activated ras and ret oncogenes induce over-expression of c-met (hepatocyte growth factor receptor) in human thyroid epithelial cells. Oncogene 1997;14: 2417–2423.
47. Croyle ML, Knauf JA, Fagin JA. RET/PTC induces EGF receptor expression and formation of RET/PTC-EGFR heterodimers in thyroid PCCL3 cells. Presented at the 75th Annual Meeting of the American Thyroid Association, 2003.
48. Santoro M, Melillo RM, Carlomagno F, et al. Molecular mechanisms of RET activation in human cancer. Ann NY Acad Sci 2002;963:116–121.
49. Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 2002; 3:383–394.
50. Santoro M, Melillo RM, Carlomagno F, et al. Molecular mechanisms of RET activation in human cancer. Ann NY Acad Sci 2002;963:116–121.
51. Grieco M, Santoro M, Berlingieri MT, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 1990;60:557–563.
52. Bongarzone I, Monzini N, Borello MG, et al. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RI alpha of cyclic AMP-dependent protein kinase A. Mol Cell Biol 1993;13:358–366.
53. Minoletti F, Butti MG, Coronelli S, et al. The two genes generating RET/PTC3 are localized in chromosomal band 10q11.2. Genes Chromosomes Cancer 1994;11:51–57.
54. Santoro M, Dathan NA, Berlingieri MT, et al. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene 1994;9:509–516.
55. Klugbauer S, Lengfelder E, Demidchik EP, et al. A new form of RET rearrangement in thyroid carcinomas of children after the Chernobyl reactor accident. Oncogene 1996;13:1099–1102.
56. Klugbauer S, Demidchik EP, Lengfelder E, et al. Detection of a novel type of RET rearrangement (PTC5) in thyroid carcinomas after Chernobyl and analysis of the involved RET-fused gene RFG5. Cancer Res 1998;58:198–203.
57. Klugbauer S, Rabes HM. The transcription coactivator HTIF1 and a related protein are fused to the RET receptor tyrosine kinase in childhood papillary thyroid carcinomas. Oncogene 1999;18:4388–4393.
58. Saenko V, Rogounovitch T, Shimizu-Yoshida Y, et al. Novel tumorigenic rearrangement, Delta rfp/ret, in a papillary thyroid carcinoma from externally irradiated patient. Mutat Res 2003;527:81–90.
59. Corvi R, Berger N, Balczon R, et al. RET/PCM-1: a novel fusion gene in papillary thyroid carcinoma. Oncogene 2000;19: 4236–4242.
60. Salassidis K, Bruch J, Zitzelsberger H, et al. Translocation t(10;14)(q11.2:q22.1) fusing the kinetin to the RET gene creates a novel rearranged form (PTC8) of the RET proto-oncogene in radiation-induced childhood papillary thyroid carcinoma. Cancer Res 2000;60:2786–2789.
61. Klugbauer S, Jauch A, Lengfelder E, et al. A novel type of RET rearrangement (PTC8) in childhood papillary thyroid carcinomas and characterization of the involved gene (RFG8). Cancer Res 2000;60:7028–7032.
62. Tong Q, Xing S, Jhiang SM. Leucine zipper-mediated dimerization is essential for the PTC1 oncogenic activity. J Biol Chem 1997;272:9043–9047.
63. Bongarzone I, Fugazzola L, Vigneri P, et al. Age-related activation of the tyrosine kinase receptor protooncogenes RET and NTRK1 in papillary thyroid carcinoma. J Clin Endocrinol Metab 1996;81:2006–2009.
64. Nikiforov YE, Rowland JM, Bove KE, et al. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res 1997;57:1690–1694.
65. Fugazzola L, Pilotti S, Pinchera A, et al. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res 1995;55:5617–5620.
66. Elisei R, Romei C, Vorontsova T, et al. RET/PTC rearrangements in thyroid nodules: studies in irradiated and not irradiated, malignant and benign thyroid lesions in children and adults. J Clin Endocrinol Metab 2001;86:3211–3216.
67. Bounacer A, Wicker R, Caillou B, et al. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene 1997;15:1263–1273.
68. Jhiang SM, Sagartz JE, Tong Q, et al. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology 1996;137:375–378.
69. Santoro M, Chiappetta G, et al. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene 1996;12: 1821–1826.
70. Powell DJJ, Russell J, Nibu K, et al. The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res 1998;58:5523–5528.
71. Viglietto G, Chiappetta G, Martinez-Tello FJ, et al. RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene 1995;11:1207–1210.
72. Sugg SL, Ezzat S, Rosen IB, et al. Distinct multiple RET/PTC gene rearrangements in multifocal papillary thyroid neoplasia. J Clin Endocrinol Metab 1998;83:4116–4122.
73. Ito T, Seyama T, Iwamoto KS, et al. In vitro irradiation is able to cause RET oncogene rearrangement. Cancer Res 1993;53: 2940–2943.
74. Mizuno T, Kyoizumi S, Suzuki T, et al. Continued expression of a tissue specific activated oncogene in the early steps of radiation-induced human thyroid carcinogenesis. Oncogene 1997; 15:1455–1460.
75. Nikiforov YE, Koshoffer A, Nikiforova M, et al. Chromosomal breakpoint positions suggest a direct role for radiation in inducing illegitimate recombination between the ELE1 and RET genes in radiation-induced thyroid carcinomas. Oncogene 1999; 18:6330–6334.
76. Nikiforova MN, Stringer JR, Blough R, et al. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 2000;290:138–141.
77. Tallini G, Santoro M, Helie M, et al. RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clin Cancer Res 1998;4:287–294.
78. Bongarzone I, Pierotti MA, Monzini N, et al. High frequency of activation of tyrosine kinase oncogenes in human papillary thyroid carcinoma. Oncogene 1989;4:1457–1462.
79. Greco A, Pierotti MA, Bongarzone I, et al. TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene 1992;7:237–242.
80. Pierotti MA, Bongarzone I, Borello MG, et al. Cytogenetics and molecular genetics of carcinomas arising from thyroid epithelial follicular cells. Genes Chromosomes Cancer 1996;16: 1–14.
81. Greco A, Mariani C, Miranda C, et al. The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol Cell Biol 1995;15:6118–6127.
82. Russell JP, Powell DJ, Cunnane M, et al. The TRK-T1 fusion protein induces neoplastic transformation of thyroid epithelium. Oncogene 2000;19:5729–5735.
83. Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658–5671.
84. Eng C. RET proto-oncogene in the development of human cancer. J Clin Oncol 1999;17:380–393.
85. Hoff AO, Cote GJ, Gagel RF. Multiple endocrine neoplasias. Annu Rev Physiol 2000;62:377–411.
86. Parma J, Duprez L, Van Sande J, et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 1993;365:649–651.
87. Duprez L, Parma J, Van Sande J, et al. Germline mutations in the thyrotropin receptor gene cause non-autoimmune autosomal dominant hyperthyroidism. Nat Genet 1994;7:396–401.
88. Parma J, Duprez L, Van Sande J, et al. Diversity and prevalence of somatic mutations in the thyrotropin receptor and Gs alpha genes as a cause of toxic thyroid adenomas. J Clin Endocrinol Metab 1997;82:2695–2701.
89. Rodien P, Ho SC, Vlaeminck V, et al. Activating mutations of TSH receptor. Ann Endocrinol (Paris) 2003;64:12–16.
90. Arturi F, Scarpelli D, Coco A, et al. Thyrotropin receptor mutations and thyroid hyperfunctioning adenomas ten years after their first discovery: unresolved questions. Thyroid 2003;13: 341–343.
91. Krohn K, Paschke R. Clinical review 133: progress in understanding the etiology of thyroid autonomy. J Clin Endocrinol Metab 2001;86:3336–3345.
92. Vanvooren V, Uchino S, Duprez L, et al. Oncogenic mutations in the thyrotropin receptor of autonomously functioning thyroid nodules in the Japanese population. Eur J Endocrinol 2002;147:287–291.
93. Mazzaferri EL. Management of a solitary thyroid nodule [see comments] [Review]. N Engl J Med 1993;328:553–559.
94. Bourasseau I, Savagner F, Rodien P, et al. No evidence of thyrotropin receptor and G(s alpha) gene mutation in high iodine uptake thyroid carcinoma. Thyroid 2000;10:761–765.
95. Cetani F, Tonacchera M, Pinchera A, et al. Genetic analysis of the TSH receptor gene in differentiated human thyroid carcinomas. J Endocrinol Invest 1999;22:273–278.
96. Russo D, Arturi F, Schlumberger M, et al. Activating mutations of the TSH receptor in differentiated thyroid carcinomas. Oncogene 1996;11:1907–1911.
97. Spambalg D, Sharifi N, Elisei R, et al. Structural studies of the thyrotropin receptor and Gs alpha in human thyroid cancers: low prevalence of mutations predicts infrequent involvement in malignant transformation. J Clin Endocrinol Metab 1996;81: 3898–3901.
98. Esapa C, Foster S, Johnson S, et al. G protein and thyrotropin receptor mutations in thyroid neoplasia. J Clin Endocrinol Metab 1997;82:493–496.
99. Russo D, Wong MG, Costante G, et al. A Val 677 activating mutation of the thyrotropin receptor in a Hurthle cell thyroid carcinoma associated with thyrotoxicosis. Thyroid 1999;9:13–17.
100. Baloch Z, LiVolsi VA. Detection of an activating mutation of the thyrotropin receptor in a case of an autonomously hyperfunctioning thyroid insular carcinoma. J Clin Endocrinol Metab 1997;82:3906–3908.
101. Cock TA, Houten SM, Auwerx J. Peroxisome proliferator-activated receptor-gamma: too much of a good thing causes harm. EMBO Rep 2004;5:142–147.
102. Kroll TG, Sarraf P, Pecciarini L, et al. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science 2000;289:1357–1360.
103. Nikiforova MN, Biddinger PW, Caudill CM, et al. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am J Surg Pathol 2002;26: 1016–1023.
104. Marques AR, Espadinha C, Catarino AL, et al. Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J Clin Endocrinol Metab 2002; 87:3947–3952.
105. Dwight T, Thoppe SR, Foukakis T, et al. Involvement of the PAX8/peroxisome proliferator-activated receptor gamma rearrangement in follicular thyroid tumors. J Clin Endocrinol Metab 2003;88:4440–4445.
106. Cheung L, Messina M, Gill A, et al. Detection of the PAX8-PPAR gamma fusion oncogene in both follicular thyroid carcinomas and adenomas. J Clin Endocrinol Metab 2003;88: 354–357.
107. Martelli ML, Iuliano R, Le PI, et al. Inhibitory effects of peroxisome poliferator-activated receptor gamma on thyroid carcinoma cell growth. J Clin Endocrinol Metab 2002;87:4728–4735.
108. Puzianowska-Kuznicka M, Krystyniak A, Madej A, Cheng SY, Nauman J. Functionally impaired TR mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab 2002;87: 1120–1128.
109. Takano T, Miyauchi A, Yoshida H, et al. Expression of TRbeta1 mRNAs with functionally impaired mutations is rare in thyroid papillary carcinoma. J Clin Endocrinol Metab 2003;88: 3447–3449.
110. Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid 2002;12:963–969.
111. Missero C, Pirro MT, Di Lauro R. Multiple ras downstream pathways mediate functional repression of the homeobox gene product TTF-1. Mol Cell Biol 2000;20:2783–2793.
112. Knauf JA, Kuroda H, Basu S, et al. RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene 2003;22: 4406–4412.
113. Medema RH, Bos JL. The role of p21ras in receptor tyrosine kinase signaling [Review]. Crit Rev Oncogenesis 1993;4:615–661.
114. Lemoine NR, Mayall ES, Wyllie FS, et al. Activated ras oncogenes in human thyroid cancers. Cancer Res 1988;48:4459–4463.
115. www.sanger.ac.uk/genetics/CGP/cosmic/, 2004.
116. Esapa CT, Johnson SJ, Kendall-Taylor P, et al. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol (Oxf) 1999;50:529–535.
117. Karga H, Lee J-K, Vickery AL, et al. Ras oncogene mutations in benign and malignant thyroid neoplasms. J Clin Endocrinol Metab 1991;73:832–836.
118. Suarez HG, du Villard JA, Severino M, et al. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene 1990;5:565–570.
119. Namba H, Rubin SA, Fagin JA. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol Endocrinol 1990;4:1474–1479.
120. Zhu Z, Gandhi M, Nikiforova MN, et al. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma: in unusually high prevalence of ras mutations. Am J Clin Pathol 2003;120:71–77.
121. Lemoine NR, Mayall ES, Wyllie FS, et al. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene 1989;4:159–164.
122. Santelli G, Franciscis V, Portella G, et al. Production of transgenic mice expressing the Ki-ras oncogene under the control of a thyroglobulin promoter. Cancer Res 1993;53:5523–5527.
123. Garcia-Rostan G, Zhao H, Camp RL, et al. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol 2003;21:3226–3235.
124. Jones CJ, Kipling D, Morris M, et al. Evidence for a telomere-independent “clock” limiting RAS oncogene-driven proliferation of human thyroid epithelial cells. Mol Cell Biol 2000;20: 5690–5699.
125. Gire V, Marshall CJ, Wynford-Thomas D. Activation of mitogen-activated protein kinase is necessary but not sufficient for proliferation of human thyroid epithelial cells induced by mutant Ras. Oncogene 1999;18:4819–4832.
126. Shirokawa JM, Elisei R, Knauf JA, et al. Conditional apoptosis induced by oncogenic ras in thyroid cells. Mol Endocrinol 2000;14:1725–1738.
127. Cheng G, Meinkoth JL. Enhanced sensitivity to apoptosis in Ras-transformed thyroid cells. Oncogene 2001;20:7334–7341.
128. Bond J, Jones C, Haughton M, et al. Direct evidence from siRNA-directed “knock down” that p16(INK4a) is required for human fibroblast senescence and for limiting ras-induced epithelial cell proliferation. Exp Cell Res 2004;292:151–156.
129. Saavedra HI, Knauf JA, Shirokawa JM, et al. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene 2000;19:3948–3954.
130. Fusco A, Berlingieri MT, Di Fiore PP, et al. One- and two-step transformations of rat thyroid epithelial cells by retroviral oncogenes. Mol Cell Biol 1987;7:3365–3370.
131. Francis-Lang H, Zannini M, De Felice M, et al. Multiple mechanisms of interference between transformation and differentiation in thyroid cells. Mol Cell Biol 1992;12:5793–5800.
132. Avvedimento VE, Musti AM, Ueffing M, et al. Reversible inhibition of a thyroid-specific trans-acting factor by Ras. Genes Dev 1991;5:22–28.
133. Gallo A, Benusiglio E, Bonapace IM, et al. v-Ras and protein kinase C dedifferentiate thyroid cells by down-regulating nuclear cAMP-dependent protein kinase A. Genes Dev 1992;6: 1621–1630.
134. Daum G, Eisenmann-Tappe I, Fries HW, et al. The ins and outs of Raf kinases. Trends Biochem Sci 1994;19:474–480.
135. Peyssonnaux C, Eychene A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell 2001;93:53–62.
136. Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet 2003;33:19–20.
137. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–954.
138. Kimura ET, Nikiforova MN, Zhu Z, et al. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 2003;63:1454–1457.
139. Trovisco V, Vieira DCI, Soares P, et al. BRAF mutations are associated with some histological types of papillary thyroid carcinoma. J Pathol 2004;202:247–251.
140. Nikiforova MN, Kimura ET, Gandhi M, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab 2003;88:5399–5404.
141. Fukushima T, Suzuki S, Mashiko M, et al. BRAF mutations in papillary carcinomas of the thyroid. Oncogene 2003;22:6455–6457.
142. Namba H, Nakashima M, Hayashi T, et al. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab 2003;88:4393–4397.
143. Xu X, Quiros RM, Gattuso P, et al. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res 2003;63:4561–4567.
144. Soares P, Trovisco V, Rocha AS, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003;22:4578–4580.
145. Cohen Y, Xing M, Mambo E, et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst 2003;95:625–627.
146. Lyons J, Landis CA, Harsh G, et al. Two G protein oncogenes in human endocrine tumors. Science 1990;249:655–659.
147. O'Sullivan CO, Barton CM, Staddon SL, et al. Activating point mutations of the gsp oncogene in human thyroid adenomas. Mol Carcinogenesis 1991;4:345–349.
148. Landis CA, Masters SB, Spada A, et al. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989;340:692–696.
149. Garcia-Rostan G, Camp RL, Herrero A, et al. Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol 2001;158:987–996.
150. Garcia-Rostan G, Tallini G, Herrero A, et al. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res 1999;59:1811–1815.
151. Miyake N, Maeta H, Horie S, et al. Absence of mutations in the beta-catenin and adenomatous polyposis coli genes in papillary and follicular thyroid carcinomas. Pathol Int 2001;51: 680–685.
152. Ishigaki K, Namba H, Nakashima M, et al. Aberrant localization of beta-catenin correlates with overexpression of its target gene in human papillary thyroid cancer. J Clin Endocrinol Metab 2002;87:3433–3440.
153. Dumont JE, Lamy F, Roger P, et al. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev 1992;72: 667–697.
154. Stergiopoulos SG, Stratakis CA. Human tumors associated with Carney complex and germline PRKAR1A mutations: a protein kinase A disease! FEBS Lett 2003;546:59–64.
155. Fagin JA. Minireview: branded from the start-distinct oncogenic initiating events may determine tumor fate in the thyroid. Mol Endocrinol 2002;16:903–911.
156. Kimura T, Van Keymeulen A, Golstein J, et al. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev 2001;22:631–656.
157. Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev 2001;12:361–373.
158. Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986;323:643–646.
159. Chen PL, Scully P, Shew JY, et al. Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 1989;58:1193–1198.
160. Dyson N, Bernards R, Friend SH, et al. Large T antigens of many polyomaviruses are able to form complexes with the retinoblastoma protein. J Virol 1990;64:1353–1356.
161. Whyte P, Buchkovich KJ, Horowitz JM, et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 1988;334:124–129.
162. Chellappan S, Kraus VB, Kroger B, et al. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci U S A 1992;89:4549–4553.
163. Huang HJ, Yee JK, Shew JY, et al. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science 1988;242:1563–1566.
164. Hu N, Gutsmann A, Herbert DC, et al. Heterozygous Rb-1 delta 20/+ mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene 1994;9: 1021–1027.
165. Harvey M, Vogel H, Lee EY, et al. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res 1995;55:1146–1151.
166. Ledent C, Franc B, Parmentier M. [Transgenic mouse models: their interest in thyroid tumors]. Arch Anat Cytol Pathol 1998;46:31–37.
167. Bartolone L, Vermiglio F, Finocchiaro MD, et al. Thyroid follicular oncogenesis in iodine-deficient and iodine-sufficient areas: search for alterations of the ras, met and bFGF oncogenes and of the Rb anti-oncogene. J Endocrinol Invest 1998; 21:680–687.
168. Zedenius J, Wallin G, Svensson A, et al. Deletions of the long arm of chromosome 10 in progression of follicular thyroid tumors. Hum Genet 1996;97:299–303.
169. Omura K, Nagasato A, Kanehira E, et al. Retinoblastoma protein and proliferating-cell nuclear antigen expression as predictors of recurrence in well-differentiated papillary thyroid carcinoma. J Clin Oncol 1997;15:3458–3463.
170. Zou M, Shi Y, Farid NR, et al. Inverse association between cyclin D1 overexpression and retinoblastoma gene mutation in thyroid carcinomas. Endocrine 1998;8:61–64.
171. Anwar F, Emond MJ, Schmidt RA, et al. Retinoblastoma expression in thyroid neoplasms. Mod Pathol 2000;13:562–569.
172. Holm R, Nesland JM. Retinoblastoma and p53 tumour suppressor gene protein expression in carcinomas of the thyroid gland. J Pathol 1994;172:267–272.
173. Jones CJ, Shaw JJ, Wyllie FS, et al. High frequency deletion of the tumour suppressor gene P16INK4a (MTS1) in human thyroid cancer cell lines. Mol Cell Endocrinol 1996;116:115–119.
174. Tung WS, Shevlin DW, Bartsch D, et al. Infrequent CDKN2 mutation in human differentiated thyroid cancers. Mol Carcinogenesis 1996;15:5–10.
175. Calabro V, Strazzullo M, La Mantia G, et al. Status and expression of the p16INK4 gene in human thyroid tumors and thyroid-tumor cell lines. Int J Cancer 1996;67:29–34.
176. Elisei R, Shiohara M, Koeffler HP, et al. Genetic and epigenetic alterations of the cyclin-dependent kinase inhibitors p15INK4b and p16INK4a in human thyroid carcinoma cell lines, and in primary thyroid cancers. Cancer 1998;83:2185–2193.
177. Schagdarsurengin U, Gimm O, Hoang-Vu C, et al. Frequent epigenetic silencing of the CpG island promoter of RASSF1A in thyroid carcinoma. Cancer Res 2002;62:3698–3701.
178. Boltze C, Zack S, Quednow C, et al. Hypermethylation of the CDKN2/p16INK4A promotor in thyroid carcinogenesis. Pathol Res Pract 2003;199:399–404.
179. Greenblatt MS, Bennett WP, Hollstein M, et al. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis [Review]. Cancer Res 1994;54:4855–4878.
180. el-Deiry WS, Harper JW, O'Connor PM, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 1994;54:1169–1174.
181. el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993;75:817–825.
182. Lane DP. Cancer. p53, guardian of the genome [News; Comment]. Nature 1992;358:15–16.
183. Donehower L, Donehower LA, Harvey M, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992;356:215–220.
184. Ito T, Seyama T, Mizuno T, et al. Genetic alterations in thyroid tumor progression: association with p53 gene mutations. Jpn J Cancer Res 1993;84:526–531.
185. Ito T, Seyama T, Mizuno T, et al. Unique association of p53 mutations with undifferentiated but not with differentiated carcinomas of the thyroid gland. Cancer Res 1992;52:1369–1371.
186. Fagin JA, Matsuo K, Karmakar A, et al. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest 1993;91:179–184.
187. Donghi R, Longoni A, Pilotti S, et al. Gene p53 et al. J Clin Invest 1993;91:1753–1760.
188. Livingstone LR, White A, Sprouse J, et al. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 1992;70:923–935.
189. Battista S, Martelli ML, Fedele M, et al. A mutated p53 gene alters thyroid cell differentiation. Oncogene 1995;11:2029–2037.
190. Fagin JA, Tang SH, Zeki K, et al. Reexpression of thyroid peroxidase in a derivative of an undifferentiated thyroid carcinoma cell line by introduction of wild-type p53. Cancer Res 1996; 56:765–771.
191. Moretti F, Farsetti A, Soddu S, et al. p53 re-expression inhibits proliferation and restores differentiation of human thyroid anaplastic carcinoma cells. Oncogene 1997;14:729–740.
192. Eng C. Genetics of Cowden syndrome: through the looking glass of oncology [Review]. Int J Oncol 1998;12:701–710.
193. Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997;16:64–67.
194. Marsh DJ, Dahia PL, Coulon V, et al. Allelic imbalance, including deletion of PTEN/MMACI, at the Cowden disease locus on 10q22–23, in hamartomas from patients with Cowden syndrome and germline PTEN mutation. Genes Chromosomes Cancer 1998;21:61–69.
195. Lynch ED, Ostermeyer EA, Lee MK, et al. Inherited mutations in PTEN that are associated with breast cancer, Cowden disease, and juvenile polyposis. Am J Hum Genet 1997;61:1254–1260.
196. Bruni P, Boccia A, Baldassarre G, et al. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: evidence that PTEN-growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene 2000;19:3146–3155.
197. Gimm O, Perren A, Weng LP, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol 2000;156:1693–1700.
198. Frisk T, Foukakis T, Dwight T, et al. Silencing of the PTEN tumor-suppressor gene in anaplastic thyroid cancer. Genes Chromosomes Cancer 2002;35:74–80.
199. Gimm O, Chi H, Dahia PL, et al. Somatic mutation and germline variants of MINPP1, a phosphatase gene located in proximity to PTEN on 10q23.3, in follicular thyroid carcinomas. J Clin Endocrinol Metab 2001;86:1801–1805.
200. Ringel MD, Hayre N, Saito J, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res 2001;61: 6105–6111.
201. Califano JA, Johns MM III, Westra WH, et al. An allelotype of papillary thyroid cancer. Int J Cancer 1996;69:442–444.
202. Ward LS, Brenta G, Medvedovic M, et al. Studies of allelic loss in thyroid tumors reveal major differences in chromosomal instability between papillary and follicular carcinomas. J Clin Endocrinol Metab 1998;83:525–530.
203. Tung WS, Shevlin DW, Kaleem Z, et al. Allelotype of follicular thyroid carcinomas reveals genetic instability consistent with frequent nondisjunctional chromosomal loss. Genes Chromosomes Cancer 1997;19:43–51.
204. Grebe SK, McIver B, Hay ID, et al. Frequent loss of heterozygosity on chromosomes 3p and 17p without VHL or p53 mutations suggests involvement of unidentified tumor suppressor genes in follicular thyroid carcinoma. J Clin Endocrinol Metab 1997;82:3684–3691.
205. Chen XN, Knauf JA, Gonsky R, et al. From amplification to gene in thyroid cancer: a high resolution mapped BAC resource for cancer chromosome aberrations guides gene discovery after comparative genome hybridization (CGH). Am J Hum Genet 1998.
206. Matsuo K, Tang SH, Fagin JA. Allelotype of human thyroid tumors: loss of chromosome 11q13 sequences in follicular neoplasms. Mol Endocrinol 1991;5:1873–1879.
207. Herrmann MA, Hay ID, Bartelt DH Jr, et al. Cytogenetic and molecular genetic studies of follicular and papillary thyroid cancers. J Clin Invest 1991;88:1596–1604.
208. Zedenius J, Wallin G, Svensson A, et al. Allelotyping of follicular thyroid tumors. Hum Genet 1995;96:27–32.
209. Eden S, Cedar H. Genomic imprinting. Action at a distance [News; Comment]. Nature 1995;375:16–17.
210. Issa JP, Vertino PM, Wu J, et al. Increased cytosine DNA-methyltransferase activity during colon cancer progression. J Natl Cancer Inst 1993;85:1235–1240.
211. Baylin SB, Herman JG, Graff JR, et al. Alterations in DNA methylation: a fundamental aspect of neoplasia [Review]. Adv Cancer Res 1998;72:141–196.
212. Herman JG, Merlo A, Mao L, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995;55:4525–4530.
213. Rideout WM3, Coetzee GA, Olumi AF, et al. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990;249:1288–1290.
214. Counts JL, Goodman JI. Alterations in DNA methylation may play a variety of roles in carcinogenesis [Review]. Cell 1995; 83:13–15.
215. Matsuo K, Tang SH, Zeki K, et al. Aberrant deoxyribonucleic acid methylation in human thyroid tumors. J Clin Endocrinol Metab 1993;77:991–995.
216. Graff JR, Greenberg VE, Herman JG, et al. Distinct patterns of E-cadherin CpG island methylation in papillary, follicular, Hurthle's cell, and poorly differentiated human thyroid carcinoma. Cancer Res 1998;58:2063–2066.
217. Xing M, Usadel H, Cohen Y, et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: a marker of malignancy and a cause of gene silencing. Cancer Res 2003;63:2316–2321.
218. Xing M, Tokumaru Y, Wu G, et al. Hypermethylation of the Pendred syndrome gene SLC26A4 is an early event in thyroid tumorigenesis. Cancer Res 2003;63:2312–2315.
219. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 1991;349:254–256.
220. Hockenbery D, Nunez G, Milliman C, et al. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990;348:334–336.
221. Castellone MD, Cirafici AM, De Vita G, et al. Ras-mediated apoptosis of PC CL 3 rat thyroid cells induced by RET/PTC oncogenes. Oncogene 2003;22:246–255.
222. Wang J, Knauf JA, Basu S, et al. Conditional expression of RET/PTC induces a weak oncogenic drive in thyroid PCCL3 cells and inhibits thyrotropin action at multiple levels. Mol Endocrinol 2003;17:1425–1436.
223. Nicolaides NC, Papadppoulos N, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994;371:75–80.
224. Parsons R, Li GM, Longley M, et al. Mismatch et al. Science 1995;268:738–740.
225. Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability [Comments]. Science 1995;268:1336–1338.
226. Nikiforov YE, Nikiforova M, Fagin JA. Prevalence of minisatellite and microsatellite instability in radiation-induced post-Chernobyl pediatric thyroid carcinomas. Oncogene 1998;17: 1983–1988.
227. Lazzereschi D, Palmirotta R, Ranieri A, et al. Microsatellite instability in thyroid tumours and tumour-like lesions. Br J Cancer 1999;79:340–345.
228. Rodrigues-Serpa A, Catarino A, Soares J. Loss of heterozygosity in follicular and papillary thyroid carcinomas. Cancer Genet Cytogenet 2003;141:26–31.
229. Bauer AJ, Cavalli LR, Rone JD, et al. Evaluation of adult papillary thyroid carcinomas by comparative genomic hybridization and microsatellite instability analysis. Cancer Genet Cytogenet 2002;135:182–186.
230. Stoler DL, Datta RV, Charles MA, et al. Genomic instability measurement in the diagnosis of thyroid neoplasms. Head Neck 2002;24:290–295.
231. Richter HE, Lohrer HD, Hieber L, et al. Microsatellite instability and loss of heterozygosity in radiation-associated thyroid carcinomas of Belarussian children and adults. Carcinogenesis 1999;20:2247–2252.
232. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997;386:623–627.
233. Jallepalli PV, Lengauer C. Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer 2001;1:109–117.
234. Tung WS, Shevlin DW, Kaleem Z, et al. Allelotype of follicular thyroid carcinomas reveals genetic instability consistent with frequent nondisjunctional chromosomal loss. Genes Chromosomes Cancer 1997;19:43–51.
235. Ward LS, Brenta G, Medvedovic M, et al. Studies of allelic loss in thyroid tumors reveal major differences in chromosomal instability between papillary and follicular carcinomas. J Clin Endocrinol Metab 1998;83:525–530.
236. Spencer F, Gerring SL, Connelly C, et al. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics 1990;124:237–249.
237. Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers [Comments]. Nature 1998;392: 300–303.
238. Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell 1997;89:727–735.
239. Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 1991;66:507–517.
240. Roberts BT, Farr KA, Hoyt MA. The Saccharomyces cerevisiae checkpoint gene BUB1 encodes a novel protein kinase. Mol Cell Biol 1994;14:8282–8291.
241. Basu J, Bousbaa H, Logarinho E, et al. Mutations in the essential spindle checkpoint gene bub1 cause chromosome missegregation and fail to block apoptosis in Drosophila. J Cell Biol 1999;146:13–28.
242. Imai Y, Shiratori Y, Kato N, et al. Mutational inactivation of mitotic checkpoint genes, hsMAD2 and hBUB1, is rare in sporadic digestive tract cancers. Jpn J Cancer Res 1999;90:837–840.
243. Gemma A, Seike M, Seike Y, et al. Somatic mutation of the hBUB1 mitotic checkpoint gene in primary lung cancer [in process citation]. Genes Chromosomes Cancer 2000;29:213–218.
244. Yamaguchi K, Okami K, Hibi K, et al. Mutation analysis of hBUB1 in aneuploid HNSCC and lung cancer cell lines. Cancer Lett 1999;139:183–187.
245. Ohshima K, Haraoka S, Yoshioka S, et al. Mutation analysis of mitotic checkpoint genes (hBUB1 and hBUBR1) and microsatellite instability in adult T-cell leukemia/lymphoma [in process citation]. Cancer Lett 2000;158:141–150.
246. Ouyang B, Knauf JA, Ain K, et al. Mechanisms of aneuploidy in thyroid cancer cell lines and tissues: evidence for mitotic checkpoint dysfunction without mutations in BUB1 and BUBR1. Clin Endocrinol (Oxf) 2002;56:341–350.
247. Pellman D. Cancer. A CINtillating new job for the APC tumor suppressor. Science 2001;291:2555–2556.
248. Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol Endocrinol 1997;11: 433–441.
249. Zhang X, Horwitz GA, Heaney AP, et al. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab 1999;84:761–767.
250. Heaney AP, Nelson V, Fernando M, et al. Transforming events in thyroid tumorigenesis and their association with follicular lesions. J Clin Endocrinol Metab 2001;86:5025–5032.
251. Yu R, Heaney AP, Lu W, et al. Pituitary tumor transforming gene causes aneuploidy and p53-dependent and p53-independent apoptosis. J Biol Chem 2000;275:36502–36505.
252. Wang Z, Yu R, Melmed S. Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol Endocrinol 2001;15: 1870–1879.
253. Boelaert K, McCabe CJ, Tannahill LA, et al. Pituitary tumor transforming gene and fibroblast growth factor-2 expression: potential prognostic indicators in differentiated thyroid cancer. J Clin Endocrinol Metab 2003;88:2341–2347.
254. Loeb LA. Cancer cells exhibit a mutator phenotype. Adv Cancer Res 1998;72:25–56.
255. Segal M, Clarke DJ. The Ras pathway and spindle assembly collide? Bioessays 2001;23:307–310.
256. Li YC, Chen CR, Chang EC. Fission yeast Ras1 effector Scd1 interacts with the spindle and affects its proper formation. Genetics 2000;156:995–1004.
257. Zecevic M, Catling AD, Eblen ST, et al. Active MAP kinase in mitosis: localization at kinetochores and association with the motor protein CENP-E. J Cell Biol 1998;142:1547–1558.
258. Shapiro PS, Vaisberg E, Hunt AJ, et al. Activation of the MKK/ERK pathway during somatic cell mitosis: direct interactions of active ERK with kinetochores and regulation of the mitotic 3F3/2 phosphoantigen. J Cell Biol 1998;142:1533–1545.
259. Denko N, Stringer J, Wani M, et al. Mitotic and post mitotic consequences of genomic instability induced by oncogenic Ha-ras. Somat Cell Mol Genet 1995;21:241–253.
260. Wani MA, Xu X, Stambrook PJ. Increased methotrexate resistance and dhfr gene amplification as a consequence of induced Ha-ras expression in NIH 3T3 cells. Cancer Res 1994;54: 2504–2508.
261. Ichikawa T, Kyprianou N, Isaacs JT. Genetic instability and the acquisition of metastatic ability by rat mammary cancer cells following v-H-ras oncogene transfection. Cancer Res 1990;50: 6349–6357.
262. Ichikawa T, Schalken JA, Ichikawa Y, et al. H-ras expression, genetic instability, and acquisition of metastatic ability by rat prostatic cancer cells following v-H-ras oncogene transfection. Prostate 1991;18:163–172.
263. de Vries JE, Kornips FH, Marx P, et al. Transfected c-Ha-ras oncogene enhances karyotypic instability and integrates predominantly in aberrant chromosomes. Cancer Genet Cytogenet 1993;67:35–43.
264. Bell B, Mazzaferri EL. Familial adenomatous polyposis (Gardner's syndrome) and thyroid carcinoma. Dig Dis Sci 1993;38: 185–190.
265. Plail RO, Bussey HJR, Glazer G, et al. Adenomatous polyposis: an association with carcinoma of the thyroid. Br J Surg 1987; 74:377–380.
266. Iwama T, Mishima Y, Utsunomiya J. The Impact of familial adenomatous polyposis on the tumorigenesis and mortality at the several organs. its rational treatment. Ann Surg 1993;217: 101–108.
267. Camiel MR, Mule JE, Alexander LL, et al. Association of thyroid carcinoma with Gardner's syndrome in siblings. N Engl J Med 1968;278:1056–1059.
268. Bell B, Mazzaferri EL. Familiar adenomatous polyposis (Gardner's syndrome) and thyroid carcinoma. A case report and review of the literature. Dig Dis Sci 1993;38:185–190.
269. Harach HR, Williams GT, Williams ED. Familial adenomatous polyposis associated thyroid carcinoma: a distinct type of follicular cell neoplasm [Review]. Histopathology 1994;25:549–561.
270. Kinzler KW, Nilbert MC, Su NKL. Identification of FAP locus genes from chromosome 5q21. Science 1991;253:661–664.
271. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589–600.
272. Cetta F, Olschwang S, Petracci M, et al. Genetic alterations in thyroid carcinoma associated with familial adenomatous polyposis: clinical implications and suggestions for early detection. World J Surg 1998;22:1231–1236.
273. Cetta F, Montalto G, Gori M, et al. Germline mutations of the APC gene in patients with familial adenomatous polyposis–associated thyroid carcinoma: results from a European cooperative study. J Clin Endocrinol Metab 2000;85:286–292.
274. Zeki K, Spambalg D, Sharifi N, et al. Mutations of the adenomatous polyposis coli gene in sporadic thyroid neoplasms. J Clin Endocrinol Metab 1994;79:1317–1321.
275. Colletta G, Sciacchitano S, Palmirotta R, et al. Analysis of adenomatous polyposis coli gene in thyroid tumours. Br J Cancer 1994;70:1085–1088.
276. Mallory SB. Cowden syndrome (multiple hamartoma syndrome). Dermatol Clin 1995;13:27–31.
277. Reed MWR, Harris SC, Quayle AR, et al. The association between thyroid neoplasia and intestinal polyps. Ann R Coll Surg Engl 1990;72:357–359.
278. Ohta S, Katsura T, Shimada M, et al. Ataxia-telangiectasia with papillary carcinoma of the thyroid. Am J Pediatr Hematol Oncol 1986;8:255–268.
279. DeLellis RA. Biology of disease. Multiple endocrine neoplasia syndromes revisited. Lab Invest 1995;72:494–505.
280. Larsson C, Skogseid B, Oberg K, et al. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988;332:85–87.
281. Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia type 1. Science 1997;276:404–407.
282. Agarwal SK, Guru SC, Heppner C, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999;96:143–152.
283. Heppner C, Bilimoria KY, Agarwal SK, et al. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene 2001; 20:4917–4925.
284. Jin S, Mao H, Schnepp RW, et al. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res 2003;63:4204–4210.
285. Sukhodolets KE, Hickman AB, Agarwal SK, et al. The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol Cell Biol 2003;23:493–509.
286. Suphapeetiporn K, Greally JM, Walpita D, et al. MEN1 tumor-suppressor protein localizes to telomeres during meiosis. Genes Chromosomes Cancer 2002;35:81–85.
287. Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 2003;113:881–889.
288. Bertolino P, Tong WM, Galendo D, et al. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol 2003; 17:1880–1892.
289. Kirschner LS, Sandrini F, Monbo J, et al. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum Mol Genet 2000;9:3037–3046.
290. Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 2000;26: 89–92.
291. Nwokoro NA, Korytkowski MT, Rose S, et al. Spectrum of malignancy and premalignancy in Carney syndrome. Am J Med Genet 1997;73:369–377.
292. Ozaki O, Ito K, Kobayashi K, et al. Familial occurrence of differentiated, nonmedullary thyroid carcinoma. World J Surg 1988;12:565–571.
293. Nemec J, Soumar J, Zamrazil V, et al. Familial occurrence of differentiated (non-medullary) thyroid cancer. Oncology 1975; 32:151–157.
294. Samaan NA. Papillary carcinoma of the thyroid: hereditary or radiation-induced? Cancer Invest 1996;7:399–400.
295. Lote K, Andersen K, Nordal E, et al. Familial occurrence of papillary thyroid carcinoma. Cancer 1980;46:1291–1297.
296. Kwok CG, McDougall IR. Familial differentiated carcinoma of the thyroid: report of five pairs of siblings. Thyroid 1995; 5:395–397.
297. Hrafnkelsson J, Tulinius H, Jonasson JG, et al. Papillary thyroid carcinoma in Iceland. Acta Oncol 1989;28:785–788.
298. Stoffer SS, Van Dyke DL, Vaden Bach J, et al. Familial papillary carcinoma of the thyroid. Am J Med Genet 1986;25:775–782.
299. Ron E, Kleinerman RA, Boice JD Jr, et al. A population-based case-control study of thyroid cancer. J Natl Cancer Inst 1987; 79:1–12.
300. Ron E, Kleinerman RA, LiVolsi VA, et al. Familial nonmedullary thyroid cancer. Oncology 1991;48:309–311.
301. Fagin JA. Familial nonmedullary thyroid carcinoma—the case for genetic susceptibility [Editorial] [Review]. J Clin Endocrinol Metab 1997;82:342–344.
302. Bignell GR, Canzian F, Shayeghi M, et al. Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am J Hum Genet 1997;61:1123–1130.
303. Canzian F, Amati P, Harach HR, et al. A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am J Hum Genet 1998;63:1743–1748.
304. Bevan S, Pal T, Greenberg CR, et al. A comprehensive analysis of MNG1, TCO1, fPTC, PTEN, TSHR, and TRKA in familial nonmedullary thyroid cancer: confirmation of linkage to TCO1. J Clin Endocrinol Metab 2001;86:3701–3704.
305. Malchoff CD, Sarfarazi M, Tendler B, et al. Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab 2000;85:1758–1764.
306. Schneider AB, Fogelfeld L. Radiation-induced endocrine tumors. Cancer Treat Res 1997;89:141–161.
307. Ron E, Lubin JH, Shore RE, et al. Thyroid cancer after exposure to external radiation: a pooled analysis of seven studies. Radiat Res 1995;141:259–277.
308. Schneider AB, Ron E, Lubin J, et al. Dose-response relationships for radiation-induced thyroid cancer and thyroid nodules: evidence for the prolonged effects of radiation on the thyroid. J Clin Endocrinol Metab 1993;77:362–369.
309. Williams D. Cancer after nuclear fallout: lessons from the Chernobyl accident. Nat Rev Cancer 2002;2:543–549.
310. Nikiforov Y, Gnepp DR, Fagin JA. Thyroid lesions in children and adolescents after the Chernobyl disaster: implications for the study of radiation carcinogenesis. J Clin Endocrinol Metab 1996;81:9.
311. Farahati J, Demidchik EP, Biko J, et al. Inverse association between age at the time of radiation exposure and extent of disease in cases of radiation-induced childhood thyroid carcinoma in Belarus. Cancer 2000;88:1470–1476.
312. Kazakov VS, Demidchik EP, Astakhova LN. Thyroid cancer after Chernobyl. Nature 1992;359:21.
313. Klugbauer S, Lengfelder E, Demidchik EP, et al. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene 1995;11:2459–2467.
314. Bounacer A, Wicker R, Caillou B, et al. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene 1997;15:1263–1273.
315. Fugazzola L, Pilotti S, Pinchera A, et al. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res 1995;55:5617–5620.
316. Rabes HM, Demidchik EP, Sidorow JD, et al. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-Chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clin Cancer Res 2000;6:1093–1103.
317. Thomas GA, Bunnell H, Cook HA, et al. High prevalence of RET/PTC rearrangements in Ukrainian and Belarussian post-Chernobyl thyroid papillary carcinomas: a strong correlation between RET/PTC3 and the solid-follicular variant. J Clin Endocrinol Metab 1999;84:4232–4238.
318. Powell DJ Jr, Russell J, Nibu K, et al. The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res 1998;58:5523–5528.
319. Nikiforov YE, Erickson LA, Nikiforova MN, et al. Solid variant of papillary thyroid carcinoma: incidence, clinical-pathologic characteristics, molecular analysis, and biologic behavior. Am J Surg Pathol 2001;25:1478–1484.
320. Nikiforov YE, Nikiforova MN, Gnepp DR, et al. Prevalence of mutations of ras and p53 in benign and malignant thyroid tumors from children exposed to radiation after the Chernobyl nuclear accident. Oncogene 1996;13:687–693.
321. Suchy B, Waldmann V, Klugbauer S, et al. Absence of RAS and p53 mutations in thyroid carcinomas of children after Chernobyl in contrast to adult thyroid tumours. Br J Cancer 1998; 77:952–955.
322. Nikiforova MN, Lynch RA, Biddinger PW, et al. RAS point mutations and PAX8-PPARgamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab 2003;88:2318–2326.