Susan E. Quaggin Jordan Kreidberg
|
|
Mammalian Kidney Development: Embryology, 3 |
|
|
|
Development of the Urogenital System, 3 |
|
|
|
Development of the Metanephros, 3 |
|
|
|
Development of the Nephron, 4 |
|
|
|
The Nephrogenic Zone, 5 |
|
|
|
Branching Morphogenesis—Development of the Collecting System, 6 |
|
|
|
Renal Stroma and Interstitial Populations, 6 |
|
|
|
Development of the Vasculature, 6 |
|
|
|
Model Systems to Study Kidney Development, 7 |
|
|
|
Organ Culture, 7 |
|
|
|
Transgenic and Knockout Mouse Models, 8 |
|
|
|
Non-mammalian Model Systems for Kidney Development, 8 |
|
|
|
Genetic Analysis of Mammalian Kidney Development, 11 |
|
|
|
Interaction of the Ureteric Bud and Metanephric Mesenchyme, 12 |
|
|
|
Formation of the Collecting System, 14 |
|
|
|
Positioning of the Ureteric Bud, 14 |
|
|
|
Molecular Biology of Nephron Development: Tubulogenesis, 15 |
|
|
|
Molecular Analysis of the Nephrogenic Zone, 15 |
|
|
|
Molecular Genetics of the Stromal Cell Lineage, 15 |
|
|
|
Molecular Genetics of Vascular Formation, 16 |
|
|
|
The Juxta-Glomerular Apparatus and the Renin-Angiotensin System, 18 |
|
|
|
Nephron Development and Glomerulogenesis, 19 |
Over the past several decades, the identification of genes and molecular pathways required for normal renal development have provided insight into our understanding of obvious developmental diseases such as renal agenesis and renal dysplasia. However, many of the genes identified have also been shown to play roles in adult-onset and acquired renal diseases such as focal segmental glomerulosclerosis. The number of nephrons present in the kidney at birth, which is determined during fetal life, predicts the risk of renal disease and hypertension later in life; a reduced number is associated with greater risk. [1] [2] [3] Discovery of novel therapeutic targets and strategies to slow and reverse kidney disease, requires an understanding of the molecular mechanisms that underlie kidney development.
MAMMALIAN KIDNEY DEVELOPMENT: EMBRYOLOGY
Development of the Urogenital System
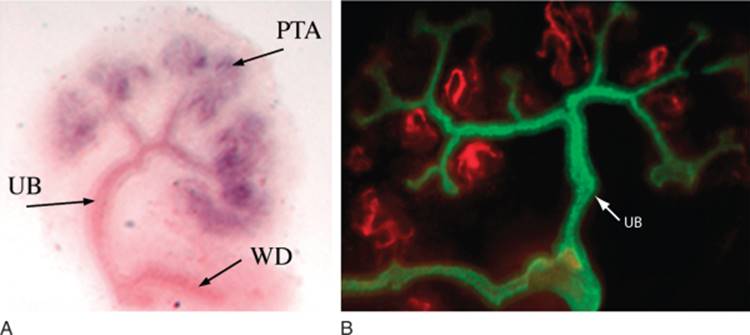
The vertebrate kidney derives from the intermediate mesoderm of the urogenital ridge, a structure found along the posterior wall of the abdomen in the developing fetus.[7] It develops in three successive stages known as the pronephros, the mesonephros, and the metanephros ( Fig. 1-1 ), although only the metanephros gives rise to the definitive adult kidney. However, earlier stages are required for development of other organs, such as the adrenal gland and gonad that also develop within the urogenital ridge. Furthermore, many of the signaling pathways and genes that play important roles in the metanephric kidney appear to play parallel roles during earlier stages of renal development, in the pronephros and mesonephros. The pronephros consists of pronephric tubules and the pronephric duct (also known as the precursor to the Wolffian duct) and develops from the rostral-most region of the urogenital ridge at 22 days of gestation (humans) and 8 days post coitum (d.p.c.; mouse). It functions in the larval stages of amphibians and fish, but not in mammals. The mesonephros develops caudal to the pronephric tubules in the mid-section of the urogenital ridge. The mesonephros becomes the functional excretory apparatus in lower vertebrates and may perform a filtering function during embryonic life in mammals. However, it largely degenerates before birth. Prior to its degeneration, endothelial, peritubular myoid, and steroidogenic cells from the mesonephros migrate into the adjacent adrenogonadal primordia, which ultimately form the adrenal gland and gonads.[10] Abnormal mesonephric migration leads to gonadal dysgenesis, a fact that emphasizes the intricate association between these organ systems during development and explains the common association of gonadal and renal defects in congenital syndromes. [11] [12] In males, production of testosterone also induces the formation of seminal vesicles, tubules of the epididymis, and portions of the vas deferens from the Wolffian duct.
|
|
|
|
|
|
|
FIGURE 1-1 Three stages of mammalian kidney development. The pronephros (P) and mesonephros (M) develop in a rostral-to-caudal direction and the tubules are aligned adjacent to the Wolffian or nephric duct (WD). The metanephros develops from an outgrowth of the distal end of the Wolffian duct known as the ureteric bud epithelium (UB) and a cluster of cells known as the metanephric mesenchyme (MM). Cells migrate from the mesonephros (M) into the developing gonad (G), which develop in close association with one another. (Adapted from Saxen L: Organogenesis of the Kidney. Cambridge, Cambridge University Press, 1987.) |
|
Development of the Metanephros
The metanephros is the third and final stage, and gives rise to the definitive adult kidney of higher vertebrates; it results from a series of inductive interactions that occur between the metanephric mesenchyme and the epithelial ureteric bud at the caudal end of the urogenital ridge. The ureteric bud (UB) is first visible as an outgrowth at the distal end of the Wolffian duct at approximately 5 weeks of gestation in humans or 10.5 days post coitus (d.p.c.) in mice. The metanephric mesenchyme (MM) becomes histologically distinct from the surrounding mesenchyme and is found adjacent to the UB. Upon invasion of UB into the MM at 11.5 d.p.c. in mice and 5 weeks in humans, signals from the MM cause the UB to branch into a T-tubule and then to undergo dichotomous branching, giving rise to the urinary collecting system and all of the collecting ducts ( Fig. 1-2 ). Simultaneously, the UB sends reciprocal signals to the MM, which is induced to condense along the surface of the bud. Following condensation, a subset of MM aggregates adjacent and inferior to the tips of the branching ureteric bud. These collections of cells are known as pre-tubular aggregates, which undergo mesenchymal-to-epithelial conversion to become the renal vesicle ( Fig. 1-3 ).
|
|
|
|
|
|
|
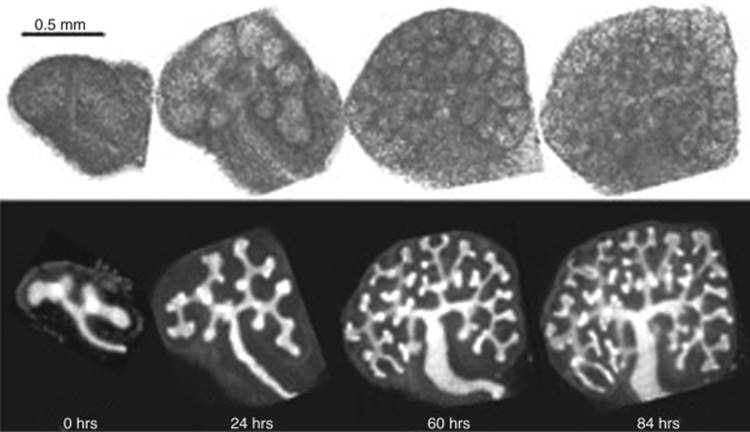
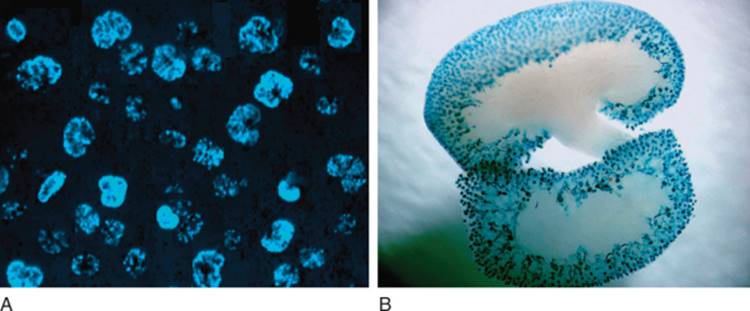
FIGURE 1-2 Organ culture of rat metanephroi dissected at T-tubule stage. Within 84 hours, dichotomous branching of the ureteric bud has occurred to provide basic architecture of the kidney. Bottom panel is stained with dolichos biflorus agglutin—a lectin specific for the UB. (Adapted from Saxen L: Organogenesis of the Kidney. Cambridge, Cambridge University Press, 1987.) |
|
|
|
|
|
|
|
|
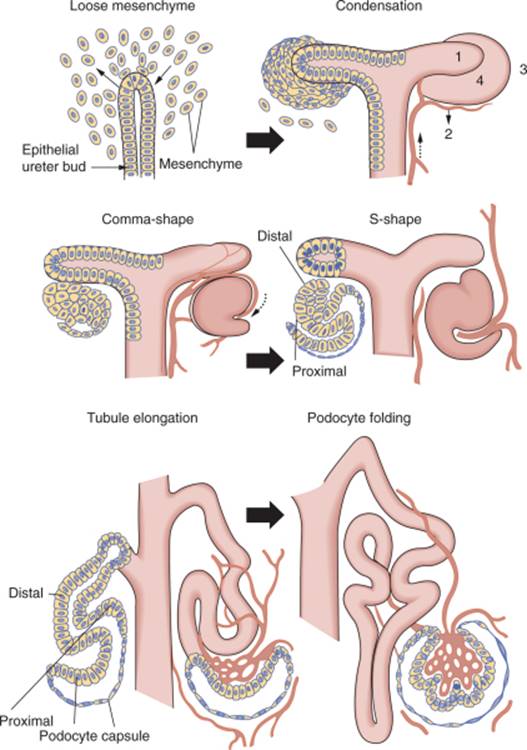
FIGURE 1-3 Schematic diagram of nephron development. As described in the text, reciprocal interaction between the ureteric bud and metanephric mesenchyme results in a series of well-defined morphologic stages leading to formation of the nephron. (From Mugrauer G, Alt FW, Ekblom P: N-myc proto-oncogene expression during organogenesis in the developing mouse as revealed by in situ hybridization. J Cell Biol 107:1325–1335, 1988. Copyright 1988, The Rockefeller University Press.) |
|
Development of the Nephron
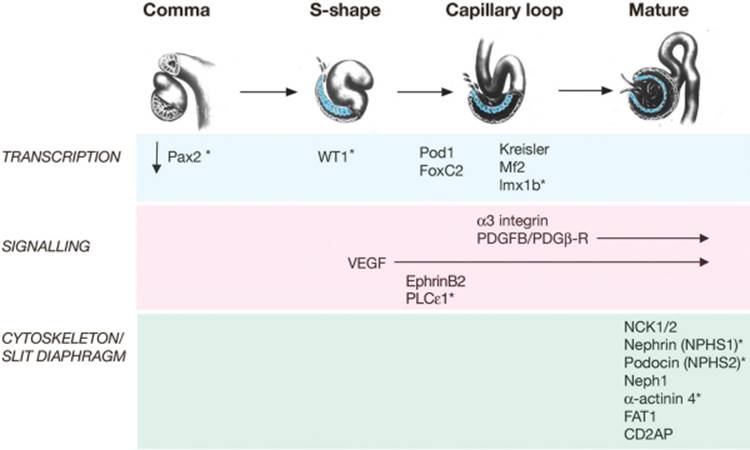
The renal vesicle segments and proceeds through a series of morphological changes to form the glomerulus and components of the tubular nephron from the proximal convoluted tubule to the distal nephron. These stages are known as comma shape, S-shape, capillary loop, and mature stage and require precise proximal-to-distal patterning and structural transformation (see Fig. 1-3 ). Remarkably, this process is repeated 600,000 to 1 million times in each developing human kidney as new nephrons are sequentially born at the tips of the UB throughout fetal life.
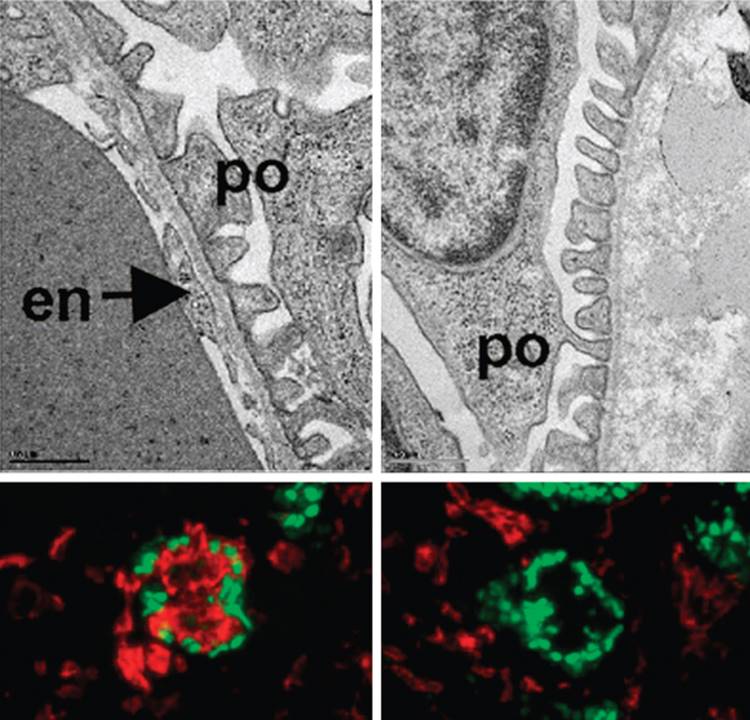
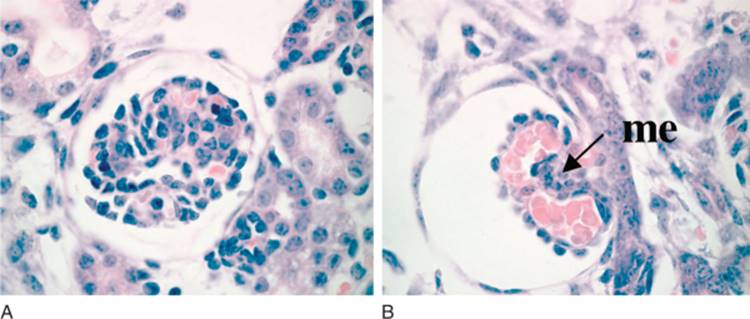
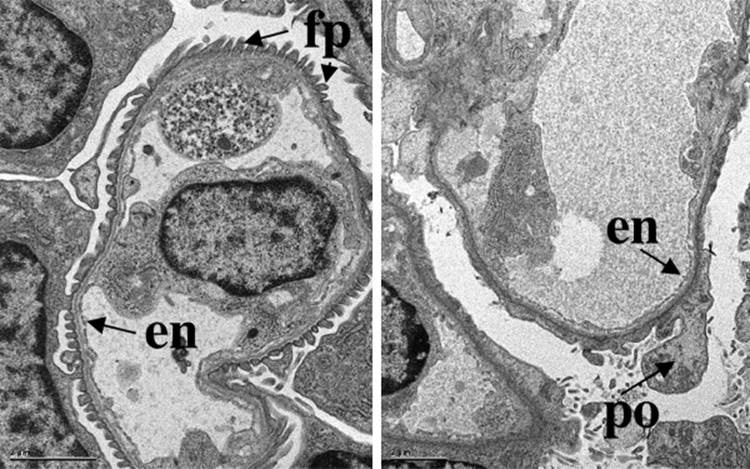
The glomerulus develops from the most proximal end of the renal vesicle that is furthest from the UB tip. [13] [14] Distinct cell types of the glomerulus can first be identified in the S-shape stage, where presumptive podocytes appear as a columnar-shaped epithelial cell layer. A vascular cleft develops and separates the presumptive podocyte layer from more distal cells that will form the proximal tubule. Parietal epithelial cells differentiate and flatten becoming Bowman's capsule, a structure that surrounds the urinary space and is continuous with the proximal tubular epithelium. Concurrently, endothelial cells migrate into the vascular cleft. Together with the glomerular visceral epithelial cells, the endothelial cells produce the glomerular basement membrane, a major component of the mature filtration barrier. Initially the podocytes are connected by intercellular tight-junctions at their apical surface.[15] As glomerulogenesis proceeds, the podocytes revert to a mesenchymal-type phenotype, flatten and spread out to cover the increased surface area of the growing glomerular capillary bed. They develop microtubular-based primary processes and actin-based secondary foot processes. During this time, the intercellular junctions become restricted to the basal aspect of the podocyte and eventually are replaced by a modified adherens-like structure known as the slit diaphragm (SD).[15] At the same time, the podocyte foot processes of adjacent cells become highly inter-digitated. The slit diaphragms function as signaling centers as well as structural components of the renal filtration apparatus that connect foot processes of adjacent podocytes and link the SD to the specialized cytoskeleton that supports foot process structure. [17] [18] [19] Mesangial cell ingrowth follows the migration of endothelial cells and is required for development and patterning of the capillary loops that are found in normal glomeruli. The endothelial cells also flatten considerably and capillary lumens are formed due to apoptosis of a subset of endothelial cells.[20] At the capillary loop stage, glomerular endothelial cells develop fenestrae, transmembrane pores that are found in semi-permeable capillary beds exposed to high flux. Positioning of the foot processes on the glomerular basement membrane and spreading of podocyte cell bodies are still incompletely understood, but share many features of synapse formation and neuronal migration. [21] [22]
In the mature stage glomerulus, the podocytes, fenestrated endothelial cells, and intervening glomerular basement membrane (GBM) comprise the filtration barrier that separates the urinary from the blood space. Together, these components provide a size- and charge-selective barrier that permits free passage of small solutes and water but prevents the loss of larger molecules such as proteins. The mesangial cells are found between the capillary loops (approximately 3 per loop); they are required to provide ongoing structural support to the capillaries and possess smooth-muscle cell-like characteristics that have the capacity to contract, which may account for the dynamic properties of the glomerulus.
The tubular portion of the nephron becomes segmented in a proximal-distal order, into the proximal convoluted tubule, the descending and ascending loops of Henle, and distal convoluted tubule. The latter portion connects to the collecting ducts, which are derived from the ureteric bud derivatives and not from the original mesenchymal component of the metanephric rudiment. Fusion events between the MM- and UB-derived portions of the nephron are required, although are poorly understood at present.
Although all segments of the nephron are present at birth and filtration occurs prior to birth, maturation of the tubule continues in the postnatal period. Increased levels of transporters, switch in transporter isoforms, alterations in paracellular transport mechanisms, and permeability and biophysical properties of tubular membranes have all been observed to occur postnatally.[23] Although additional studies are needed, these observations emphasize the importance of considering developmental stage of the nephron in interpretation of renal transport and may explain the age of onset of symptoms in inherited transport disorders; some of these issues may be recapitulated in acute renal injury.
The Nephrogenic Zone
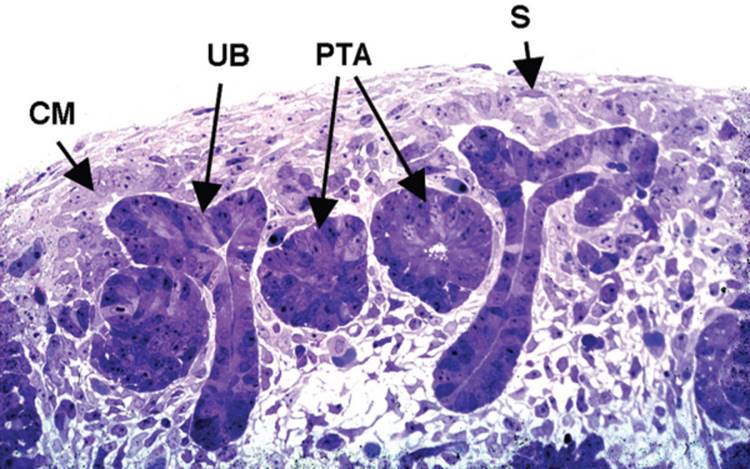
After the first few rounds of branching of the ureteric bud derivatives, and the concomitant induction of nephrons from the mesenchyme, the kidney begins to become divided between an outer cortical region where nephrons are being induced, and an inner medullary region where the collecting system will form. As growth continues, successive groups of nephrons are induced at the peripheral regions of the kidney, known as the nephrogenic zone ( Fig. 1-4). Thus, within the developing kidney, the most mature nephrons are found in the innermost layers of the cortex, and the most immature nephrons in the most peripheral regions. At the extreme peripheral lining, under the renal capsule, a process that appears to nearly exactly recapitulate the induction of the original nephrons can be observed, where numerous ureteric bud-like structures are inducing areas of condensed mesenchyme. Indeed, whether there are significant molecular differences between the induction of the original nephrons and these subsequent inductive events is not known. Also unknown is whether there exists a stem-like population of cells within or adjacent to the nephrogenic zone. It is apparent from the histology of the nephrogenic zone that the mesenchyme condensed around these derivatives of the ureteric bud must continually replenish itself, as well as provide a substrate for the induction of successive rounds of nephrons. However, it is not known whether there is a small subset of cells that have the stem-like properties of self-renewal and differentiation, or whether these properties apply to the whole population of condensed mesenchyme present in the nephrogenic zone.
|
|
|
|
|
|
|
FIGURE 1-4 The nephrogenic zone. As described in the text, nephrons are continually produced in the nephrogenic zone throughout fetal life. CM, condensing mesenchyme; UB, ureteric bud; PTA, pretubular aggregate; S, stromal cell lineage (spindle-shaped cells). |
|
Branching Morphogenesis—Development of the Collecting System
The collecting system is composed of hundreds of tubules through which the filtrate produced by the nephrons is conducted out of the kidney and to the ureter and then the bladder. Water and salt absorption and excretion, NH3 transport and H+ ion secretion required for acid-base homeostasis also occur in the collecting ducts, under different regulatory mechanisms, and using different transporters and channels than are active in the tubular portions of the nephron. The collecting ducts are all derived from the original ureteric bud. So, whereas each nephron is an individual unit separately induced and originating from a distinct pretubular aggregate, the collecting ducts are the product of branching morphogenesis from the ureteric bud. Consi-derable remodeling is involved in forming collecting ducts from branches of ureteric bud, and how this occurs remains incompletely understood.[24] The branching is highly patterned, with the first several rounds of branching being somewhat symmetrical, followed by additional rounds of asymmetric branching, in which a main trunk of the collecting duct continues to extend towards the nephrogenic zone, while smaller buds branch as they induce new nephrons within the nephrogenic zone. Originally, the ureteric bud derivatives are branching within a surrounding mesenchyme. Ultimately, they form a funnel-shaped structure in which a cone-shaped grouping of ducts or papilla sits within a funnel or calyce that drains into the ureter. The mouse kidney has a single papilla and calyce, whereas a human kidney has 8 to 10 papillae, each of which drains into a minor calyce, with several minor calyces draining into a smaller number of major calyces.
Renal Stroma and Interstitial Populations
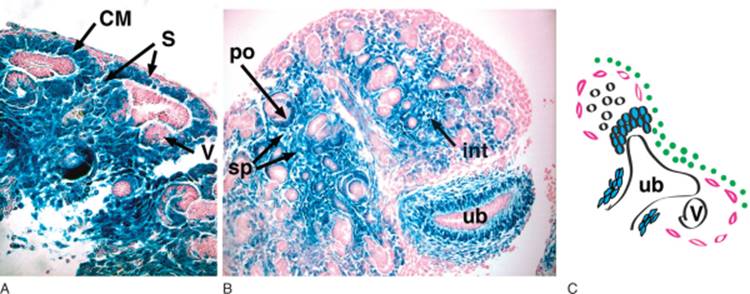
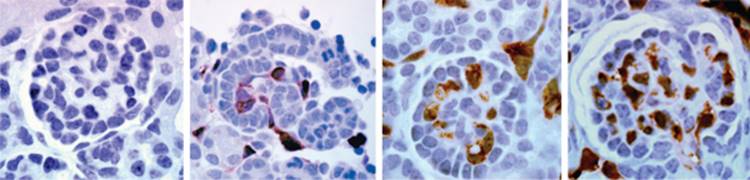
For decades in classic embryologic studies of kidney development, emphasis has been placed on the reciprocal inductive signals between MM and UB. However, in recent years, interest in the stromal cell as a key regulator of nephrogenesis has arisen. [14] [25] [26] [27] Stromal cells also derive from the metanephric mesenchyme, but are not induced to condense by the UB. Two distinct populations of stromal cells have been described: cortical stromal cells exist as a thin layer beneath the renal capsule while medullary stromal cells populate the interstitial space between the collecting ducts and tubules (see Fig. 1-8 ). Cortical stromal cells also surround the condensates and provide signals required for ureteric bud branching and patterning of the developing kidney. Disruption or loss of these stromal cells leads to failure of UB branching, a reduction in nephron number, and disrupted patterning of nephric units with failure of cortical-medullary boundary formation. A reciprocal signaling loop from the UB exists to properly pattern stromal cell populations. Loss of these UB-derived signals leads to a buildup of stromal cells beneath the capsule that are several layers thick. As nephrogenesis proceeds, stromal cells differentiate into peritubular interstitial cells and pericytes that are required for vascular remodeling, and production of extracellular matrix responsible for proper nephric formation. These cells migrate from their position around the condensates to areas between the developing nephrons within the medulla. Although stromal cells derive from MM, it is not yet clear if MM that give rise to stromal cell and nephric lineages derive from the same progenitor cell or a different cell.
|
|
|
|
|
|
|
FIGURE 1-8 Populations of cells within the metanephric mesenchyme. As described in the text, these populations are defined by morphologic and molecular characteristics. Metanephroi from a 14.5 d.p.c (A) and 15.5 d.p.c. (B) Pod1/lacZ mouse are stained with lacZ. Pod1-expressing cells stain blue. Stromal cells (S; pink in C) are seen surrounding condensing mesenchyme (CM). Metanephrogenic population (green in C) remain unstained. By 15.5 d.p.c. a well-developed interstitial compartment is seen and consists of peritubular fibroblasts, medullary fibroblasts, and pericytes. Loose and condensed mesenchymal cells are also observed around the stalk of the ureteric bud in B. v, renal vesicle; po, podocyte precursors; sp, stromal pericytes; int, interstitium. C, Schematic diagram of mesenchymal populations include the metanephrogenic precursors (in green), uninduced mesenchyme (white), condensing mesenchyme around the UB tips and stalk (blue) and stromal cell lineage (pink). (Reproduced with permission from Developmental Dynamics.) |
|
Development of the Vasculature
The microcirculations of the kidney include the specialized glomerular capillary system responsible for production of the ultrafiltrate and the vasa rectae, peritubular capillaries involved in the countercurrent mechanism. In the adult, each kidney receives 10% of the cardiac output.
Vasculogenesis and angiogenesis have been described as two distinct processes in blood vessel formation. The first refers to de novo differentiation of previously nonvascular cells into structures that resemble capillary beds, whereas angiogenesis refers to sprouting from these early beds to form mature vessel structures including arteries, veins, and capillaries. Both processes are involved in development of the renal vasculature. At the time of UB invasion (11 d.p.c.; all timing given is for mice), the MM is avascular but by 12 d.p.c. a rich capillary network is present and by 14 d.p.c. vascularized glomeruli are present. Transplantation experiments support a model whereby endothelial progenitors within the MM give rise to renal vessels in situ,[28] although the origin of large blood vessels is still debated. At 13 d.p.c., capillaries form networks around the developing nephric tubules and by 14 d.p.c., the hilar artery and first-order interlobar renal artery branches can be identified. These branches will form the cortico-medullary arcades; and interlobular arteries that branch from these arcades. Further branching produces the glomerular afferent arterioles. From 13.5 d.p.c. onward, endothelial cells migrate into the vascular cleft of developing glomeruli, where they undergo differentiation to form the glomerular capillary loops. The efferent arterioles carry blood away from the glomerulus to a system of fenestrated peritubular capillaries that are in close contact with the adjacent tubules and receive filtered water and solutes reabsorbed from the filtrate. These capillaries have few pericytes. In comparison, the vasa recta, which surround the medullary tubules and are involved in urinary concentration are also fenestrated but have more pericytes. They arise from the efferent arterioles of deep glomeruli. The peritubular capillary system surrounding the proximal tubules is well developed in the late fetal period, whereas the vasa rectae mature 1 to 3 weeks postnatally.
MODEL SYSTEMS TO STUDY KIDNEY DEVELOPMENT
Organ Culture
The Kidney Organ Culture System: Classical Studies
Metanephric kidney organ culture ( Fig. 1-5 ) formed the basis for extensive classical studies of embryonic induction. Parameters of induction such as the temporal and physical constraints on exposure of the inductive tissue to the mesenchyme were determined, as were the time periods during which various tubular elements of the nephron were first observed in culture.
|
|
|
|
|
|
|
FIGURE 1-5 Metanephric organ explants. A, In situ analysis for Pax2 that marks pretubular aggregates (PTA) and the ureteric (UB) and Wolffian duct (WD). B, Immunohistochemical stain for proximal tubular cell brush border (red) and pan cytokeratin (green) marks the developing nephrons and ureteric bud, respectively. |
|
Mutant Phenotypic Analyses
As originally shown by Grobstein, Saxen, and colleagues in classical studies of embryonic induction, the two major components of the metanephric kidney, the mesenchyme and the ureteric bud, could be separated from each other, and the isolated mesenchyme could be induced to form nephron-like tubules by a selected set of other embryonic tissues, the best example of which is embryonic neural tube. [7] [29] This phenomenon can be distinguished from placing the whole metanephric rudiment, including the ureteric bud, in culture, in that when the whole rudiment is placed in culture, there is induction of nephrons, branching of the ureteric bud, and continued growth of the rudiment. In contrast, when neural tube is used to induce the separated mesenchyme, there is terminal differentiation of the mesenchyme into tubules, but not significant tissue expansion. The isolated mesenchyme experiment has proven useful in the analysis of renal agenesis phenotypes, where there is no outgrowth of the ureteric bud. In these cases the mesenchyme can be placed in contact with neural tube to determine whether it has the intrinsic ability to differentiate. Most often, when the renal agenesis is due to the mutation of a transcription factor, tubular induction is not rescued by neural tube, as could be predicted for transcription factors, which would be expected to act in a cell-autonomous fashion.[6] In the converse situation, in which renal agenesis is caused by loss of a gene function in the ureteric bud, such as EMX-2, it is usually possible for embryonic neural tube to induce tubule formation in isolated mesenchymes.[30] Therefore the organ culture induction assay can be used to test hypotheses concerning whether a particular gene is required in the mesenchyme or ureteric bud. Recently, as chemical inhibitors specific for various signal transduction pathways have been synthesized and become available, it has been possible to add these to organ cultures and observe effects that are informative about the roles of specific pathways in development of the kidney. Examples are the use of MAP kinase inhibitors and inhibitors of the Notch signaling pathway.
Anti-Sense Oligonucleotides and siRNA in Organ Culture
Several studies have described the use of antisense oligonucleotides and more recently, siRNA molecules, to inhibit gene expression in kidney organ culture. Among the earliest of these was the inhibition of the low affinity nerve growth factor receptor, p75 or NGFR, by anti-sense oligonucleotides,[31] a treatment that decreased the growth of the organ culture. A subsequent study could not duplicate this phenotype,[32] though there were possible differences in experimental techniques.[33] An additional study using anti-sense oligonucleotides to Pax2 also showed this gene to be crucial in the mesenchymal to epithelial transformation. [8] [9] More recently, one report has demonstrated that siRNA to the WT1 and Pax2 genes can inhibit early nephron differentiation.[34]
Organ Culture Microinjection
A novel approach to the organ culture system has also yielded insights as to a possible function of the WT1 gene in early kidney development. A system was established to microinject and electroporate DNA plasmid expression constructs into the condensed mesenchyme of organ cultures.[35] The results with this system are described in the section on Wt1.
Transgenic and Knockout Mouse Models
Over the past two decades, the generation and analysis of knockout and transgenic mice have provided tremendous insight into kidney development ( Table 1-1 ). [36] [37] Although homologous recombination to delete genes within the germline also known as standard “knockout” technology has provided information about the biological functions of many genes in kidney development, several disadvantages exist. Disruption of gene function in embryonic stem (ES) cells may result in embryonic or perinatal lethality, precluding the functional analysis of the gene in the kidney that develops relatively late in fetal life. Additionally, many genes are expressed in multiple cell types, and the resulting knockout phenotypes can be complex and difficult or impossible to dissect. The ability to limit gene targeting to specific renal cell types overcomes some of these problems and the temporal control of gene expression permits more precise dissection of a gene's function. A number of mouse lines exist that may be used to target specific kidney cell lineages ( Table 1-2 ; Fig. 1-6 ). As with any experimental procedure, numerous caveats exist that the investigator must take into account in interpretation of data (reviewed in Refs 38, 39); these include determining the completeness of excision at the locus of interest, the timing and extrarenal expression of the promoters, and general toxicity of expressed proteins to the cell of interest. In spite of these caveats, they remain a powerful tool. The next generation of targeting includes improved efficiency using BAC targeting approaches, siRNA and microRNA approaches, and large genome-wide targeting efforts already underway at many academic and pharmaceutical institutions.
TABLE 1-1 -- Summary of Knockout and Transgenic Models for Kidney Development
|
Kidney Phenotype |
Mouse (Knockout or Mutation) Other Affected Organs |
Human (Naturally Occurring Mutation) |
References |
|
Aplasia (Variable) |
|||
|
WT-1 |
Gonad, mesothelium, heart, lung |
Wilms tumor, WAGR, Denys-Drash |
4–6 [4] [5] [6] |
|
Pax-2 |
Genital tract, gonad |
Renal hypoplasia, VUR, and optic nerve colobomas |
8, 9 [8] [9] |
|
Pax-2/Pax-8 |
Defect in intermediate mesoderm transition, failure of pronephric duct formation |
|
|
|
Emx-2 |
Genital tract, gonad |
30 |
|
|
Lim-1 |
Genital tract, gonads, anterior head |
66 |
|
|
Hox-A11/D11 |
Distal limbs, vas deferens |
217 |
|
|
Retinoic acid receptor αγ/αβ2 |
Skeleton, many visceral abnormalities including renal hypoplasia, dysplasia |
12, 14, 16 [12] [14] [16] |
|
|
GDNF, c-ret, GRFα1 |
UB failure, enteric neurons |
Hirschsprung disease |
67–69, 173–177 [67] [68] [69] [173] [174] [175] [176] [177] |
|
Integrin-α8 |
Reduced UB branching |
87 |
|
|
Danforth Short Tail |
Short tail, UB failure |
218 |
|
|
KAL mutation |
|
Kallman syndrome (olfactory bulb agenesis) |
|
|
Heparan sulfate 2-sulfotransferase |
Lack of UB branching and mesenchymal condensation |
|
219 |
|
EYA-1 (Eyes absent-1) |
Branchio-oto-renal syndrome (branchial fistulae, deafness) |
62, 74 [62] [74] |
|
|
Six1 |
Branchio-oto-renal syndrome |
64 |
|
|
Gremlin |
|
71 |
|
|
Sal1 |
Severe renal dysplasia/renal agenesis |
Townes-Brock syndrome (anal, renal, limb, ear anomalies) |
65, 178 [65] [178] |
|
Dysplasia/Hypoplasia/Low Nephron Mass |
|||
|
FoxD1 (BF-2) |
Reduced UB branching/stromal patterning defects |
|
26 |
|
BMP-7 |
Reduced MM survival |
86 |
|
|
Wnt-4 |
Failure of MM induction |
84, 179 [84] [179] |
|
|
AP-2 |
MM failure, craniofacial and skeletal defects |
220 |
|
|
Cyclooxygenase-2 |
Oligonephronia |
221 |
|
|
Lmx-1β |
Renal dysplasia, skeletal abnormalities |
Nail-patella syndrome |
160, 180 [160] [180] |
|
FGF-7 |
Small kidneys, reduction in nephron number |
91 |
|
|
Increased Branching |
|||
|
Slit2/robo2 |
Increased branching of UB |
|
102 |
|
Cysts |
|||
|
KIF3A |
Polycystic kidney disease (tubular-selective) |
|
181 |
|
HNF1β |
182 |
||
|
VHL |
Renal cysts (tubular-selective) |
|
183 |
|
Peroxisomal assembly factor-1 |
Zellweger syndrome |
OMIM*214100 |
|
|
Bcl-2 |
Renal hypoplasia and cysts |
|
|
|
MKS1 |
Meckel syndrome (multicystic dysplasia, neural tube defect) |
222 |
|
|
PKD1, PKD2 |
Renal cysts |
AD PKD |
184 |
|
Later phenotypes (Glomerular, vascular, glomerular basement membrane) |
|||
|
PDGFB/PDGFR-β |
Lack of mesangial cells, ballooned glomerular capillary loop |
|
157, 158 [157] [158] |
|
MPV-17 |
Nephrotic syndrome |
223 |
|
|
Integrin-α3 |
Reduced UB branching, glomerular defects, poor foot process formation, lung |
98 |
|
|
CD151 |
Focal segmental glomerulosclerosis, massive proteinuria, disorganized GBM, tubular cystic dilation |
End stage kidney failure, regional skin blistering, sensorineural deafness |
228 |
|
|
Alport syndrome |
|
185, 186 [185] [186] |
|
187 |
|||
|
188 |
|||
|
Intracerebral hemorrhage and strokes |
189 |
||
|
Lamb2 |
Proteinuria prior to the onset of foot process effacement |
|
190, 191 [190] [191] |
|
Lama5 |
Defective glomerulogenesis, abnormal GBM, poor podocyte adhesion, loss of mesangial cells |
192 |
|
|
Lama5;Mr51 |
Ballooned capillary loop, proteinuria |
193 |
|
|
Lama5;Mr5G2 |
Nephrotic syndrome |
194 |
|
|
Agrin |
No glomerular permeability defect (podocyte-selective) |
224 |
|
|
Perlecan heparan sulfated sites |
No baseline defects; proteinuria with albumin loading |
195 |
|
|
Entactin-1 |
Abnormal GBM |
196 |
|
|
Angiotensin II type-2 receptor |
Various collecting system defects |
CAKUT syndrome |
143, 144, 147 [143] [144] [147] |
|
Eagle-Barrett (prune belly) syndrome |
|
(-) Abdominal wall musculature, VUR, cryptorchidism |
|
|
BMP-4 (heterozygous) |
Renal hypoplasia/dysplasia, hydroureter, ectopic uterovesical junction |
89 |
|
|
Foxc1 (Mfl) |
Renal duplication, multiple ureters, hydroureter/hydronephrosis |
101 |
|
|
Mf2 |
Small kidneys with few nephrons |
197 |
|
|
Glypican-3 |
Disorganized tubules and medullary cysts |
Simpson-Golabi-Behmel syndrome |
198–201 [198] [199] [200] [201] |
|
Notch2 |
Lack of glomerular endothelial and mesangial cells |
103, 104 [103] [104] |
|
|
Pod1/tcf21 |
Lung and cardiac defects, sex reversal and gonadal dysgenesis, vascular defects, disruption in UB branching, impaired podocyte differentiation, dilated glomerular capillary, poor mesangial migration |
11, 111 [11] [111] |
|
|
FoxC2 |
Impaired podocyte differentiation, dilated glomerular capillary loop, poor mesangial migration |
49 |
|
|
Kreisler (maf-1) |
Abnormal podocyte differentiation |
159 |
|
|
Nephrin |
Absent slit diaphragms |
Congenital nephrosis of the Finnish variety |
162 |
|
Neph 1 |
Abnormal slit diaphragm function, FSGS |
48 |
|
|
Podocin |
Congenital nephrosis, FSGS, vascular defects |
Steroid-resistant FSGS |
166, 202 [166] [202] |
|
PLCε1 |
Diffuse mesangial sclerosis; FSGS |
225 |
|
|
GNE/MNK (M712T) |
Hyposialation defect, foot process effacement, GBM splitting, proteinuria and hematuria |
Hereditary inclusion body myopathy |
226 |
|
FAT1 |
Foot process fusion, failure of foot process formation |
203 |
|
|
NCK1/2 |
Failure of foot process formation (podocyte-selective) |
17 |
|
|
CD2AP |
FSGS, immunotactoid nephropathy |
169 |
|
|
Alpha-actinin 4 |
Glomerular developmental defects, FSGS |
AD FSGS |
167, 168 [167] [168] |
|
VEGF-A |
Endotheliosis, disruption of glomerular filtration barrier formation, nephrotic syndrome (podocyte-selective) |
118, 119 [118] [119] |
|
|
Angiopoietin2 |
Cortical peritubular capillary abnormalities |
131 |
|
|
ILK1 |
Nephrotic syndrome (podocyte-selective) |
204 |
|
|
VHL |
RPGN (podocyte-selective) |
137 |
|
|
VUR, vesicoureteral reflux; UB, ureter bud; MM, metanephric mesenchyme; AD, autosomal dominant; PKD, polycystic kidney disease; VHL, von Hippel-Lindau; GBM, glomerular basement membrane; FSGS, focal segmental glomerulosclerosis; RPGN, rapidly progressive glomerulonephritis. |
TABLE 1-2 -- Conditional Mouse Lines for the Kidney
|
Promoter |
Renal Expression |
Extrarenal Expression |
Reference |
|
Kidney androgen promoter 2 |
Proximal tubules |
Brain |
[205] |
|
γ-Glutamyl transpeptidase |
Cortical tubules |
None |
[206] |
|
Na/glucose cotransporter (SGLT2) |
Proximal tubules |
None |
[207] |
|
PEPCK |
Proximal tubules |
Liver |
[183] |
|
Aquaporin-2 |
Principal cells of collecting duct |
Testis, vas deferens |
[208] |
|
Hox-B7 |
Collecting ducts, Ureteric bud, Wolffian bud, ureter |
Spinal cord, dorsal root ganglia |
[209] |
|
Ksp-cadherin |
Renal tubules, collecting ducts, ureteric bud, Wolffian duct, mesonephros |
Müllerian duct |
[210] |
|
Tamm-Horsfall protein |
Thick ascending limbs of loops of Henle |
Testis, brain |
[211] |
|
Nephrin |
Podocytes |
Brain |
212, 213 [212] [213] |
|
Podocin |
Podocytes |
None |
[214] |
|
Renin |
Juxtaglomerular cells, afferent arterioles |
Adrenal gland, testis, sympathetic ganglia, etc. |
[141] |
|
FoxD1/BF2 |
Stromal cells |
? |
|
|
Six2 |
Metanephric mesenchyme |
? |
|
|
Pax3 |
Metanephric mesenchyme |
Neural tube, neural crest |
215, 216 [215] [216] |
|
|
|
|
|
|
|
FIGURE 1-6 Glomeruli expressing cyan fluorescent protein (A) or beta galactosidase (B). Transgenic mice were generated using the nephrin-promoter to direct expression of either CFP or beta-galactosidase specifically to developing and mature podocytes. |
|
In contrast to gene targeting experiments where the gene is known at the beginning of the experiment (reverse genetics), random mutagenesis represents a complimentary phenotype-driven approach (forward genetics). Random mutations are introduced into the genome at high efficiency by chemical or “gene-trap” mutagenesis. Consecutively, large numbers of animals are screened systematically for specific phenotypes of interest. As soon as a phenotype is identified, test breeding is used to confirm the genetic nature of the trait. The mutated gene is then identified by chromosomal mapping and positional cloning. There are two major advantages to genome-wide based approaches compared to reverse genetics: (1) most knockouts lead to major gene disruptions, which may not be relevant to the subtle gene alterations that underlie human renal disease; (2) many of the complex traits underlying congenital anomalies and acquired diseases of the kidney are unknown, making predictions about the nature of the genes that are involved in these diseases difficult.
One of the most powerful and well-characterized mutagens in the mouse is the chemical mutagen, N-ethyl-N-nitrosourea (ENU). It acts through random alkylation of nucleic acids inducing point mutations in spermatogonial stem cells of injected male mice. [40] [41] This results in multiple point mutations within the spermatogonia of the male, who is then bred to a female mouse of different genetic background. Resulting F1 offspring are screened for renal phenotypes of interest (e.g., dysplastic, cystic) and heritability. Mutations may be complete or partial loss-of-function, gain-of-function, or altered function and can be dominant or recessive. The specific locus mutation frequency of ENU is 1 in 1000. Assuming a total number of 25,000 to 40,000 genes in the mouse genome, a single treated male mouse should have between 25 and 40 different heterozygous mutagenized genes. In the case of multigenic phenotypes, segregation of the mutations in the next generation allows the researcher to focus on monogenic traits. In each generation, 50% of the mutations are lost, and only the mutation underlying the selected phenotype is maintained in the colony. A breeding strategy that includes backcrossing to the female genetic strain enables rapid mapping of the ENU mutation that occurred on the male genetic background.
The screening in ENU-mutagenesis experiments can focus on dominant or recessive renal mutations. Screening for dominant phenotypes is popular as breeding schemes are simple and a great amount of mutants can be recovered through this approach. About 2% of all F1 mice display a heritable phenotypic abnormality. [42] [43] A number of large ENU mutagenesis projects are now underway, with mutant strains available to interested researchers. It is possible to design “sensitized screens” on a smaller scale, which increases the ability to identify genes in a pathway of interest. For example, in renal glomerular development, the phenotype of a genetic mouse strain with a tendency to develop congenital nephrosis (e.g., CD2AP haploinsufficiency[44]) may be enhanced or suppressed by breeding to a mutagenized male. The modifier gene may then be mapped using the approach outlined earlier. This approach has been successfully used to identify genes involved in neural development, [45] [46] but has not yet been exploited to full potential by the renal community.
Other genome-wide approaches that have led to the discovery of novel genes in kidney development and disease include gene trap consortia, [47] [48] and transcriptome/proteome projects.[49] The interested reader is referred to the following web site: www.cmhd.on.ca![]() .
.
Non-mammalian Model Systems for Kidney Development
Organisms separated by millions of years of evolution from humans, still provide useful models to study the genetic basis and function of mammalian kidney development. This stems from the fact that all of these organisms possess excretory organs designed to remove metabolic wastes from the body, and that genetic pathways involved in other aspects of invertebrate development may serve as templates to dissect pathways in mammalian kidney development. In support of the latter argument, elucidation of the genetic interactions and molecular mechanism of the Neph1 ortholog and nephrin-like molecule—SYG1 and SYG2—in synapse formation in C. Elegans is providing major clues to the function of these genes in glomerular and slit diaphragm formation and function in mammals.[50]
The excretory organs of invertebrates differ greatly in their structure and complexity and range in size from a few cells in C. elegans, to several hundred cells in the Malpighian tubules of Drosophila, to the more recognizable kidneys in amphibians, birds, and mammals. In the soil nematode, C. elegans, the excretory system consists of a single large H-shaped excretory cell, a pore cell, a duct cell, and a gland cell. [51] [52] C. elegans provides many benefits as a model system: the availability of powerful genetic tools including “mutants by mail”, a short life and reproductive cycle, a publicly available genome sequence and resource database (www.wormbase.org![]() ), the ease of performing genetic enhancer-suppressor screens in worms and the fact that they share many genetic pathways with mammals. Major contributions in our understanding of the function of polycystic and cilia-related genes have been made from studying C. elegans. The PKD1 and PKD2 homologs, LOV1 and LOV2, are involved in cilia development and function of the mating organ required for mating behavior. [53] [54] Strides in understanding the function of the slit diaphragm have also been made from C. elegans as described earlier.
), the ease of performing genetic enhancer-suppressor screens in worms and the fact that they share many genetic pathways with mammals. Major contributions in our understanding of the function of polycystic and cilia-related genes have been made from studying C. elegans. The PKD1 and PKD2 homologs, LOV1 and LOV2, are involved in cilia development and function of the mating organ required for mating behavior. [53] [54] Strides in understanding the function of the slit diaphragm have also been made from C. elegans as described earlier.
In Drosophila, the “kidney” consists of Malpighian tubules that develop from the hindgut and perform a secretion reabsorption filtering function.[55] They express a number of mammalian gene homologues (e.g., Cut, members of the Wingless pathway) that have subsequently been shown to play major roles in mammalian kidney development. Furthermore, studies on myoblast fusion and neural development in Drosophila—two processes that may not appear to be related to kidney development at first glance—have provided major clues into development and function of slit diaphragms.[56] Mutations in the Neph ortholog Irregular chiasm C-roughest (IrreC-rst) are associated with neuronal defects and abnormal patterning of the eye. [57] [58]
The pronephros, which is only the first of three stages of kidney development in mammals, is the final and only kidney of jawless fish, whereas the mesonephros is the definitive kidney in amphibians. The pronephros found in larval stage zebra fish consists of two tubules connected to a fused, single, midline glomerulus. The zebrafish pronephric glomerulus expresses many of the same genes found in mammalian glomeruli including VEGFA, NPHS1, NPHS2, Wt1 and contain podocytes and fenestrated endothelial cells.[59] Advantages to the zebra fish as a model system include its short reproductive cycle, transparency of the larvae with easy visualization of defects in pronephric development without sacrificing the organism, availability of the genome sequence, the ability to rapidly knockdown gene function using morpholino oligonucleotides, and the ability to perform functional studies of filtration using fluorescently tagged labels of varying sizes.[60] These features lend the zebra fish to both forward and reverse genetic screens and currently, there are several labs performing knockdown screens of mammalian homologues in zebra fish and genome-wide mutagenesis screens to study renal function.
The pronephros of Xenopus has also been used as a simple model to study early events in nephrogenesis. Similar to the fish, the pronephros consists of a single glomus, paired tubules, and a duct. The fact that Xenopus embryos develop rapidly outside the body (all major organ systems are formed by 6 days of age), the ease of injecting DNA, mRNA, and protein and ability to perform grafting and in vitro culture experiments establish the frog as a valuable model system to dissect early inductive and patterning cues.[61]
GENETIC ANALYSIS OF MAMMALIAN KIDNEY DEVELOPMENT
Much has been learned about the molecular genetic basis of kidney development over the past 15 years. This understanding has primarily been gained through the phenotypic analysis of mice carrying targeted mutations that affect kidney development. Additional information has been gained by identification and study of genes that are expressed in the developing kidney, even though the targeted mutation, or “knockout”, either has not yet been done, or the knockout has not affected kidney development or function. In this section, we categorize the genetic defects based on the major phenotype and stage of disrupted development. It must be emphasized that many genes are expressed at multiple time stages of renal development and may play pleiotropic roles that are not yet entirely clear.
Interaction of the Ureteric Bud and Metanephric Mesenchyme
The molecular analysis of the initiation of metanephric kidney development has included a series of classical experiments using organ culture systems that allow separation of the ureteric bud and metanephric mesenchyme, and more recently, the analysis of many gene targeted mice whose phenotypes have included various degrees of renal agenesis. The organ culture system has been in use since the seminal experiments, beginning in the 1950s, of Grobstein, Saxen, and colleagues. These experiments showed that the induction of the mesenchymal to epithelial transformation within the mesenchyme required the presence of an inducing agent, provided by the ureteric bud. The embryonic neural tube was found to be able to substitute for the epithelial bud, and experiments involving the placement of the inducing agent on the opposite side of a porous filter from the mesenchyme provided information about the degree of contact required between them. A large series of experiments using the organ culture provided information about the timing of appearance of different proteins normally observed during the induction of nephrons, and the time intervals that were crucial in maintaining contact between the inducing agent and the mesenchyme to obtain induction of tubules.
The work with the organ culture system provided an extensive framework on which to base further studies of organ development, and remains in extensive use to this day. However, the modern era of studies on the early development of the kidney began with the observation of renal agenesis phenotypes in gene targeted or knockout mice, the earliest among these, the knockout of several transcription factors including the Wilms' Tumor-1 gene, also known as WT-1,[6] Pax-2,[9] Eya-1,[62] Six-1, [63] [64] Sall-1,[65] Lim-1,[66] and Emx-2.[30] The knockout of several secreted signaling molecules such as GDNF, [67] [68] [69] GDF-11,[70] Gremlin[71] or their receptors, including c-Ret[72] and GFRa-1[73] also resulted in renal agenesis, at least in the majority of embryos.
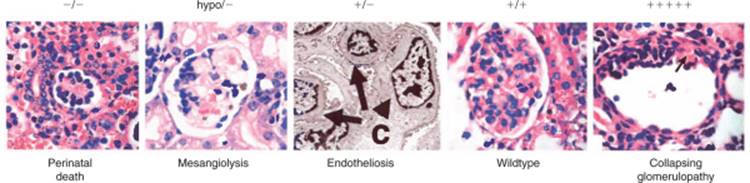
Renal Agenesis Phenotypes from Transcription Factor Mutations
In embryos with the phenotype of renal agenesis, the most common observation is for there to be a histologically distinct patch of mesenchyme located in the normal location of the metanephric mesenchyme, but for there to be no outgrowth of the ureteric bud. An exception is the Eya-1 mutant embryo, where this distinct patch of mesenchyme is not found, suggesting that Eya-1 expression may indeed be the earliest determinant of the metanephric mesenchyme yet identified ( Fig. 1-7 ). Together, the phenotypes of these knockout mice have provided an initial molecular hierarchy of early kidney development. There is evidence for at least three major pathways involved in determining the appearance and early function of the metanephric mesenchyme. One is the Eya-1/Six-1 pathway, which has also been implicated in kidney development through the study of humans with urogenital defects. Eya1 and Six1 mutations are found in humans with branchio-oto-renal (BOR) syndrome.[74] It is now known, through in vitro experiments, that Eya1 and Six1 form a regulatory complex that appears to be involved in transcriptional regulation. [75] [76] Interestingly, a phosphatase activity is associated with this complex.[76] Moreover, Eya and Six family genes are co-expressed in several tissues in mammals, Xenopus and Drosophila, further supporting a functional interaction of these genes. [62] [63] [64] [77] [78] Direct transcriptional targets of this complex appear to include the pro-proliferative factor c-Myc.[76] In the Eya1-deficient urogenital ridge, it has recently been demonstrated that, unlike with some other renal agenesis phenotypes, there is no histologically distinct group of cells in the normal location of the metanephric mesenchyme.[79] Consistent with this finding, Six1 is either not expressed or highly diminished in expression in the location of the metanephric mesenchyme of Eya1 -/- embryos. [76] [77] [78] [79] These findings may identify Eya1 as a gene involved in early commitment of this group of cells to the metanephric lineage. Although Six1 and Eya1 may act in a complex together, the Six1 phenotype is somewhat different, in that a histologically distinct mesenchyme is present at E11.5, without an invading ureteric bud, similar to the other renal agenesis phenotypes. [63] [64] Eya1 is expressed in the Six1-/- mesenchyme, suggesting that Eya1 is upstream of Six1. Additionally, Sal1 and Pax2 are not expressed in the Six1 mutant mesenchyme, though Wt1 is expressed. [63] [64] [79] (There are discrepancies in the literature about Pax2 expression in Six1 mutant embryos, which may reflect the exact position along the anterior-posterior axis of the urogenital ridge of Six1 mutant embryos from which sections are obtained.)
|
|
|
|
|
|
|
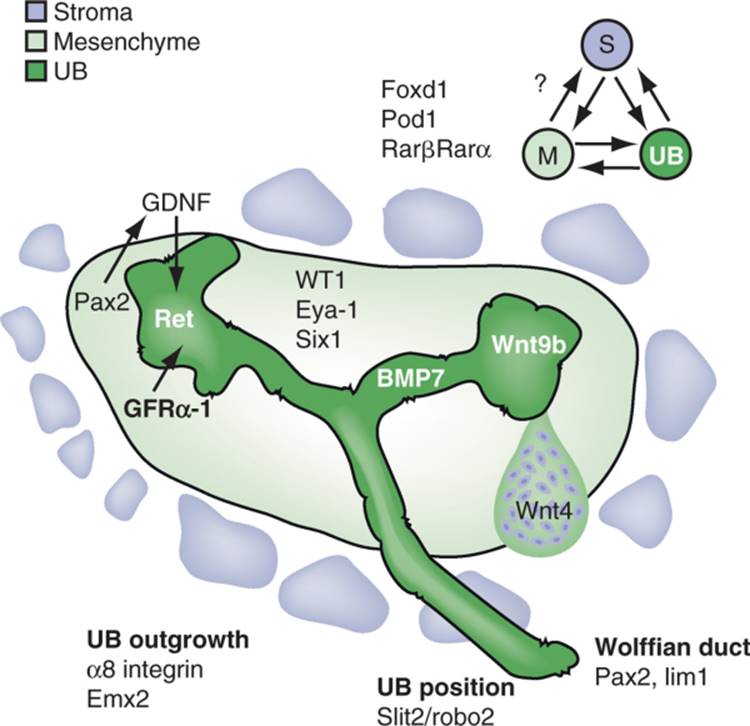
FIGURE 1-7 Reciprocal interactions occur between all three major compartments of the metanephros (stroma, mesenchyme, and ureteric bud (UB)). Pretubular aggregate is shown on right side. Selected molecules that play key roles in these interactions are shown for simplicity. The Pax2/GDNF loop underlies a push to UB branching whereas the Wnt side represents induction of nephrons. |
|
A second pathway in the metanephric mesenchyme involves Pax-2 and GDNF. In Pax2 -/- embryos, Eya1, Six1 and Sal1 are expressed,[79] suggesting that the Eya-1/Six-1 pathway is not downstream, but may be upstream of Pax-2. Through a combination of molecular and in vivo studies, it has been demonstrated that Pax2 appears to act as a transcriptional activator of GDNF,[80] the major growth factor attracting and maintaining outgrowth of the ureteric bud and its derivative branches.
The third major pathway involves WT-1 and VEGF-A.[35] A novel approach to the organ culture system involving microinjection and electroporation has also yielded insights as to a possible function of the WT1 gene in early kidney development. Over-expression of WT1 from an expression construct led to high-level expression of vascular endothelial growth factor-A (VEGF-A). The target of VEGF-A appeared to be Flk-1 (VEGFR-2) expressing angioblasts at the periphery of the mesenchyme. Blocking signaling through Flk-1, if done when the metanephric rudiment was placed in culture, blocked expression of Pax2 and GDNF, and consequently the continued branching of the ureteric bud and induction of nephrons by the bud. Addition of the Flk-1 blockade after the organ had been in culture for 48 hours had no effect, indicating that the angioblast-derived signal was required to initiate kidney development, but not to maintain continued development. The signal provided by the angioblasts is not yet known, nor is it known whether WT1 is a direct transcriptional activator of VEGF-A. Flk-1 signaling is also known to be required to initiate hepatocyte differentiation during liver development.
Genes Required by the Ureteric Bud in Early Kidney Development
Genes expressed by the ureteric bud are also crucially involved in the inductive events of early kidney development. Examples include the transcription factor Emx-2 and c-Ret, the receptor for GDNF. C-Ret is a receptor tyrosine kinase, and presumably transduces signals to the epithelial cells of the bud that result in continued branching and proliferation. Interestingly, the failure of ureteric bud growth and branching of the GDNF homozygous mutant embryos can be rescued in situations in which the embryo also carries a transgene that specifically directs GDNF expression in the ureteric bud, and not in the mesenchyme. This autocrine-like rescue of ureteric bud growth and branching indicates that the pattern of branching is not determined by a specific pattern of GDNF expression in the mesenchyme; rather, any local source of GDNF elicits the usual pattern of branching.
Signaling Factors in Early Kidney Development
The signaling pathways described in the metanephric mesenchyme were identified by mutation of transcription factors. Presumably these transcription factors direct the expression of genes that encode proteins that act within the cell, in addition to genes encoding secreted molecules that act to convey signals from one group of cells to an adjacent or nearby group of cells. As previously mentioned, it has been demonstrated that Pax2 regulates the expression of GDNF, and WT1 regulates VEGF-A. In the case of other signaling molecules, it has yet to be determined what transcription factors control their expression in different cell types within the early kidney. Nevertheless, several groups of signaling molecules have been identified to be of great importance in early kidney development.
Fibroblast growth factors (FGFs) have been implicated in the very early stages of differentiation of the nephron. Conditional mutation of fibroblast growth factor receptors in the mesenchyme results in renal agenesis with a ureteric bud, with expression of early markers such as Eya1 and Six1 in the vicinity of the ureteric bud, but without expression of slightly later markers such as Six2 or Pax2, and no branching of the bud or induction of nephrons.[81] Two groups have published conditional mutations in the FGF8 gene, which eliminate expression of FGF8 in the mesenchyme of the early kidney. [82] [83] Failure to properly express FGF8 did not block formation of a WT1 and Pax2-expressing condensed mesenchyme, but Wnt4-expressing pretubular aggregates were not present, and consequently S-shaped bodies, the precursor of the nephron, never developed. [82] [83] Interestingly, these conditionally mutant kidneys were smaller with fewer branches of the collecting ducts, suggesting that nephron differentiation may have a role in driving continued branching morphogenesis of the collecting system.
Two members of the Wnt family of signaling molecules are expressed by the ureteric bud, Wnt 11 and Wnt 9b. The Wnt family was originally discovered as the Wingless mutation in Drosophila, and in mammals as genes found at retroviral integration sites in mammary tumors in mice. Wnt11 is expressed at the tips of the buds, and decreased branching is observed in its absence, though there is no specific effect on the induction of the mesenchymal to epithelial transformation. In contrast, Wnt9b, which is expressed in the entire ureteric bud except the very tips, appears to be the vital molecule expressed by the bud that induces the mesenchyme. In the absence of Wnt 9b, the bud merges from the Wolffian duct and invades the mesenchyme, which condenses around the bud, but pretubular aggregates do not form, and there is no mesenchymal to epithelial transformation. No further branching of the bud is observed. Thus, Wnt 9b is the closest candidate identified to date, which is likely to be the crucial molecule produced by the bud that stimulates induction of the nephrons.
A third member of the Wnt family, Wnt 4, is expressed in the pretubular aggregate, and is required for the mesenchymal to epithelial transformation.[84] In Wnt4 mutant embryos, pre-tubular aggregates are present but they fail to undergo the mesenchymal to epithelial transformation into the tubular precursor of the mature nephron, indicating a role for Wnt4 in the formation of epithelial cells from mesenchyme.[84] The role of Wnt signaling has been studied in vitro, by exposing isolated metanephric mesenchymes to fibroblast cultures transfected with Wnt-expressing DNA vectors. Several Wnt's were found to be able to induce the mesenchymal to epithelial transformation, similar to that observed using embryonic neural tube. In considering these experiments in light of the more recently published Wnt 9b phenotype, it is worth considering whether the neural tube induction experiment can be viewed as a recapitulation of the Wnt 4 or Wnt 9b function, or both. As previously noted, a distinction between the induction by the ureteric bud versus neural tube, is that the bud stimulates both proliferation and mesenchymal to epithelial transformation, whereas the neural tube, or Wnt 4 expression by fibroblasts, only stimulates differentiation, without significant proliferation. On the other hand, both the classical neural tube experiment and the Wnt-expressing fibroblast experiment seemingly bypass a step normally observed in kidney development, that of formation of the pretubular aggregate inferior to the tips of the bud. Instead, aggregates form randomly within the isolated mesenchyme. Therefore, the neural tube rescue experiments are more consistent with a Wnt 4 rather than a Wnt 9b function. Further experimentation will be needed, however, to determine whether Wnt 9b, in the absence of the bud, can stimulate proliferation in addition to differentiation, in order to determine whether the expression of Wnt 9b is the major criterion that distinguishes the induction by the bud from induction by neural tube.
The bone morphogenetic protein (BMP) family of proteins is an additional family of secreted signaling proteins that plays a crucial role in the developing kidney.
Bmp-7 is first expressed in the ureteric bud, and then in the condensed mesenchyme. [85] [86] In the absence of Bmp7, the first round of nephrons are induced, but there is no further kidney development. [85] [86] It has been suggested that this first round of nephrons might result from maternal contribution of Bmp7 across the placenta, and it is not known whether Bmp7 is absolutely required for the induction of nephrons.
Adhesion Proteins in Early Kidney Development
A current theme in cell biology is that growth factor signaling often occurs coordinately with signals from the extracellular matrix transduced by adhesion receptors such as members of the integrin family. α8β1 integrin is expressed by cells of the metanephric mesenchyme,[87] which binds a novel molecule named nephronectin,[88] expressed on the ureteric bud. In most α8 integrin mutant embryos, the ureteric bud arrests its outgrowth upon contact with the metanephric mesenchyme.[87] In a small portion of embryos, this block is overcome, and a single, usually hypoplastic, kidney develops. Thus, the interaction of α8β1 integrin with nephronectin must have an important role in the continued growth of the ureteric bud into the mesenchyme. This phenotype also implies that there is something about the interaction of ureteric bud cells with the metanephric mesenchyme, which distinguishes it from the interaction of the ureteric bud with the undifferentiated mesenchyme of the urogenital ridge, through which it must briefly pass before encountering the metanephric mesenchyme. Whether α8β1 integrin:nephronectin signaling occurs in concert with growth factor signaling is not yet known.
Formation of the Collecting System
Formation of the collecting system is the result of the branching morphogenesis of the ureteric bud and its derivative branches, followed by extensive remodeling of those initial branches, to finally form the papillary region of the medulla, as well as the collecting ducts within the cortex and outer medulla. The overall structure of the kidney is largely patterned by the collecting system, and understanding the pathways that drive the formation of the collecting system will be an essential component of understanding how the kidney derives its overall structure.
The molecular events crucial to the development of the collecting system occur largely as interactions between the mesenchymal cells and the epithelial derivatives of the ureteric bud. This is especially true in the early phases of kidney development, when the epithelial branches of the bud are surrounded by mesenchyme. At later stages of development, when the cortex and medulla form distinct areas of the kidney, and nephrons and stromal cells compose much of the cortex, it is presently much less clear what the important cellular interactions are, even when it is known what molecules are important.
Several families of secreted growth factors have been demonstrated to be important in the patterning of the collecting system, including members of the BMP,[89] sonic hedgehog,[90] and FGF families.[91] The role of GDNF, required for initial outgrowth of the bud and to drive continued branching, was previously discussed. Conditional gene targeting of FGF receptor 2 (but not of FGFR1) in the ureteric bud results in greatly decreased branching of the bud.[92] Mice carrying a mutation of FGF7 have smaller collecting systems,[91] though this phenotype is not as severe as the conditional mutation of FGFR2, implying that additional FGFs are probably involved in branching of the ureteric bud. The role of FGF8 in the mesenchyme, with regard to nephron development, was previously discussed. Whether FGF8 or another FGF is also driving development of the collecting system is not clear, but it is interesting to note that mutations that block induction of nephrons also tend to eliminate further branching and growth of the derivative branches of the ureteric bud.
The role of BMP7 in early kidney development has been discussed in a previous section. Two other prominent members of the BMP family, BMP2 and 4, also have significant roles in formation of the collecting system,[89] which were more difficult to decipher, as mouse embryos carrying mutations in these factors undergo embryonic demise too early to identify an effect on kidney development. In organ culture, BMP7 stimulates branching, whereas BMP2 was found to inhibit branching of the derivatives of the ureteric bud. [93] [94] Further study of the role of BMP2 utilized a constitutively active form of the BMP2 receptor, ALK3, specifically expressed in the derivatives of the ureteric bud. This resulted in medullary dysplasia, resembling medullary cystic disease observed in humans.[95] It appears that BMP2 signaling normally acts to suppress a proliferation signal mediated by smad signaling and β-catenin, which acts to stimulate expression of the pro-proliferative transcription factor c-Myc.[96]
Diminished branching is also observed in kidneys of sonic hedgehog (shh)-deficient embryos.[90] This phenotype bears resemblance to the renal dysplasia observed in humans with mutations of the Gli3 gene, which encodes an effector of the Shh signaling pathway.[97] Increased expression of Pax2 and Sal1, required for normal kidney development, as well as cell cycle regulatory genes N-myc and cyclin D1, were observed in Shh-deficient kidneys.[97]Interestingly, the Shh-deficient kidney phenotype was rescued by inhibiting signaling through Gli3, providing genetic confirmation that Gli3 is a regulator of the Shh pathway.[97]
Targeted mutation in mice of the α3 integrin gene, discussed later in regard to its role in glomerular development, also results in a poorly formed papilla, with fewer collecting ducts and increased interstitium.[98] α3β1 integrin is expressed in the ureteric bud and collecting ducts.[99] In vitro, α3β1 integrin appears to have a role both in cell-matrix and cell-cell interaction,[100] but the latter role has not been verified in vivo, though α3β1 integrin is expressed basolaterally in developing tubules, consistent with both roles. As integrins are known to signal in coordination with growth factor receptors, it will be of interest to determine if α3β1 integrin is involved in any of the signaling pathways discussed previously in this section.
Positioning of the Ureteric Bud
A final aspect of kidney development that is of great relevance to renal and urological congenital defects in humans relates to the positioning of the ureteric bud. Incorrect position of the bud, or duplication of the bud, results in abnormally shaped kidneys, and/or incorrect insertion of the ureter into the bladder, and resultant ureteral reflux that can pre-dispose to infection and scarring of the kidneys and urological tract.
FoxC1 is a transcription factor of the Forkhead family, expressed in the intermediate mesoderm and the metanephric mesenchyme adjacent to the Wolffian duct. In the absence of Foxc1, the expression of GDNF adjacent to the Wolffian duct is less restricted than in wild-type embryos, and there are resultant ectopic ureteric buds, giving rise to duplex ureters, one of which is a hydroureter, and to hypoplastic kidneys.[101] Additional molecules that regulate the location of ureteric bud outgrowth are slit2 and ROBO2, signaling molecules best known for their role in axon guidance in the developing nervous system. Slit is a secreted factor, and ROBO is its receptor. Slit2 is mainly expressed in the Wolffian duct, whereas Robo2 is expressed in the mesenchyme.[102] In embryos deficient in either Slit2 or ROBO2, there are ectopic ureteric buds, similar to the Foxc1 mutant. (Dissimilar to the Foxc1 phenotype was the observation that none of the ureters in Slit2/ROBO2 mutants failed to undergo the normal remodeling that results in insertion in the bladder; instead, the ureters remained connected to the nephric duct.[102]) The domain of GDNF expression is expanded anteriorly in the absence of either Slit2 or ROBO2. The expression of Pax2, Eya1, and Foxc1, all thought to regulate GDNF expression, was not dramatically different in the absence of Slit2 or ROBO2, suggesting that Slit/ROBO signaling was not upstream of these genes. It is possible that Slit/ROBO signaling is regulating the point of ureteric bud outgrowth, by regulating the GDNF expression domain downstream of Pax2 or Eya1. An alternative explanation is that Slit/ROBO are acting independently of GDNF, and that the expanded GDNF domain is a response to, rather than a cause of, ectopic ureteric buds.
Molecular Biology of Nephron Development: Tubulogenesis
While gene targeting and other analyses have identified many genes involved in the initial induction of the metanephric kidney and the formation of the pre-tubular aggregate, much less is presently known about how the pre-tubular aggregate develops into the mature nephron, a process through which a simple tubule elongates, convolutes, and differentiates into multiple distinct segments with different functions. In considering how this segmentation occurs, it has been considered whether there will be similarities to other aspects of development, such as the limb or neural tube, where there is segmentation along various axes.
The Notch group of signaling molecules has been implicated in directing segmentation of the nephron. Notch family members are transmembrane proteins whose cytoplasmic domains are cleaved by the γ-secretase enzyme, upon the interaction of the extracellular domain with transmembrane ligand proteins of the delta and jagged families, found on adjacent cells.[103] Thus, Notch signaling occurs between adjacent cells, in contrast to signaling by secreted growth factors, which may occur at a distance from the growth factor-expressing cells. The cleaved portion of the Notch cytoplasmic domain translocates to the nucleus, where it has a role in directing gene expression. Mice homozygous for a hypomorphic allele of Notch2 have abnormal glomeruli, with a failure to form a mature capillary tuft. [104] [105] Because null mutants of notch family members usually result in early embryonic lethality, further analysis of Notch family function in kidney development has made use of the organ culture model. When metanephric rudiments are cultured in the presence of a γ-secretase inhibitor, [106] [107] there is diminished expression of podocyte and proximal tubule markers, in comparison with distal tubule markers and branching of the ureteric bud. When the γ-secretase inhibitor is removed, there seems to be a better recovery of expression of proximal tubule markers, in comparison with markers of podocyte differentiation. Similar results were observed in mice carrying targeted mutation of the PSEN1 and PSEN2 genes that encode a component of the γ-secretase complex.[108] These results, while requiring confirmation from mice carrying conditional mutations in notch genes themselves, suggest that Notch signaling is involved in patterning the proximal tubule and glomerulus. Similar results were obtained from mice with mutations in the gene encoding γ-secretase.
There is one example so far of a transcription factor being involved in the differentiation of a specific cell type in the kidney. The phenotype is actually found in the collecting ducts, rather than in the nephron itself, but is discussed in this section as it is demonstrative of the types of phenotypes it is expected will be found as additional mutant mice are examined. Two cell types are normally found in the collecting ducts—principal cells, which mediate water and salt reabsorption, and intercalated cells that mediate acid-base transport. In the absence of the Foxi1 transcription factor, there is only one cell type present in collecting ducts, and many acid-base-transport proteins normally expressed by intercalated cells are absent.[109]
Molecular Analysis of the Nephrogenic Zone
The molecular biology of the nephrogenic zone remains largely to be explored, especially that pertaining to whether there exists a population of kidney stem cells. As noted, the histology of this zone resembles the early developing kidney, with condensed mesenchyme, which expresses Pax2 and low levels of Wt1, surrounding ureteric bud-like structures. Unknown is how this population of condensed mesenchyme is maintained; whether it self-replenishes, such that it could be regarded as a stem-like population, or whether there is a subset of cells within the condensed mesenchyme that are the stem-like cells. Alternatively, there may be a population of cells, not within the condensed mesenchyme itself, which both self-replenishes and gives rise to successive populations of condensed mesenchyme in each successive layer of the nephrogenic zone. At present, no molecular markers have been identified that distinguish a subset of cells that might be a stem-like population, apart from the condensed mesenchyme as a whole.
Molecular Genetics of the Stromal Cell Lineage
In recent years, a key role for the stromal cell lineage in kidney development was discovered largely through the analysis of knockout mice. FoxD1 (formerly BF2) is a winged helix transcription factor; in the kidney it is only expressed in stromal cells that are found in a rim beneath the renal capsule and as a layer of cells surrounding the mesenchymal condensates.[26] Despite the restricted distribution of FoxD1-positive cells, major defects in the development of adjacent renal tubules and glomeruli were observed in FoxD1 KO mice. These results demonstrate that stromal cells are required for nephrogenesis and furthermore that the model of reciprocal signaling between the UB and condensates must be extended to include the stromal cell compartment[14] ( Fig. 1-8 ).
Pod1 (tcf21/capsulin/epicardin), a member of the basic-helix-loop-helix family of transcription factors, is also expressed in the stromal cell lineage, as well as in condensing MM. [110] [111] Pod1 is also expressed in a number of differentiated renal cell types that derive from these mesenchymal cells and include developing and mature podocytes of the renal glomerulus, cortical and medullary peritubular interstitial cells, pericytes surrounding small renal vessels, and adventitial cells surrounding larger blood vessels. The defect in nephrogenesis observed in Pod1 KO mice is similar to the defect seen in BF2 knockout mice, with disruption of branching morphogenesis and an arrest and delay in glomerulogenesis and tubulogenesis. Analysis of chimeric mice that are derived from Pod1 KO embryonic stem cells and GFP-expressing embryos, demonstrated both cell autonomous and non-cell-autonomous roles for Pod1 in nephrogenesis.[112] Most strikingly, the glomerulogenesis defect is rescued by the presence of wild-type stromal cells (i.e., mutant cells will epithelialize and form nephrons normally as long as they are surrounded by wild-type stromal cells in keeping with the model outlined in Figs. 1-7 and 1-8 [7] [8]). In addition, there is a cell autonomous requirement for Pod1 in stromal mesenchymal cells to allow differentiation into interstitial cell and pericyte cell lineages of the cortex and medulla as Pod1 null ES cells were unable to contribute to these populations.
Although many of the defects in the Pod1 mutant kidneys phenocopy those seen in the BF2 mutant kidneys, there are important differences. In the kidneys of Pod1 KO mice, there are vascular anomalies and absence of pericyte differentiation that were not reported in FoxD1 mutant mice. These differences might result from the broader domain of Pod1 expression, which also includes the condensing mesenchyme, podocytes and medullary stromal cells, in addition to the stromal cells that surround the condensates. In contrast to FoxD1, Pod1 is not highly expressed in the thin rim of stromal cells found immediately beneath the capsule, suggesting that FoxD1 and Pod1 might mark early and late stromal cell lineages, respectively, with overlap in the stroma that surrounds the condensates.[27] However, definitive co-labeling studies to address this issue have not been performed. As both Pod1 and FoxD1 are transcription factors it is interesting to speculate that they might interact or regulate the expression of a common stromal “inducing factor”.
Vitamin A deficiency has been associated with a variety of birth defects, including renal dysplasia; vitamin A is the ligand for retinoic acid receptors (RARs) including RARA and RARB2, both of which are expressed in the stromal cell lineage. Mice that lack both of these receptors and thus have decreased vitamin A signaling, demonstrate decreased branching of the UB, patterning defects in the stromal cell lineage with a buildup of stromal cell layers beneath the capsule and defects in nephron patterning. [14] [27] [113] Transgenic overexpression of the tyrosine kinase receptor, c-ret, under the regulation of a ureteric bud-specific promoter from the Hoxb7 gene rescued the observed defects although retinoic acid treatment alone could not. Taken together, these results show that a vitamin A dependent signal is required in stromal cells for UB branching and that UB branching is required to pattern the stroma.
Molecular Genetics of Vascular Formation
Vasculogenesis and angiogenesis both contribute to vascu-lar development within the kidney. Endothelial cells may be identified through the expression of the tyrosine kinase signaling receptor, VEGFR –2 (flk1/KDR).[114] Reporter mouse strains that carry lacZ or GFP cDNA cassettes “knocked into” the VEGFR-2 locus, permit precise snapshots of vessel development, as all the vascular progenitor and differentiated cells in these organs express a blue or green color ( Fig. 1-9 ). Use of other knock-in strains allows identification of endothelial cells lining arteriolar or venous vessels.[115]
|
|
|
|
|
|
|
FIGURE 1-9 Developing glomeruli stained with an antibody to the GFP (green fluorescent protein). Control glomerulus from a wild-type mouse. S-shape, capillary and mature glomeruli in an 18.5 d.p.c. metanephros from a Flk1-GFP mouse strain. All endothelial cells express the GFP protein that is expressed under control of the endogenous Flk1/VEGFR-2 promoter. (Reproduced with permission from the Journal of American Society of Nephrology.) |
|
Over the past decade, a number of growth factors and their receptors have been identified that are required for vasculogenesis and angiogenesis. Gene deletion studies in mice have shown that VEGF-A and its cognate receptor VEGFR-2 are essential for vasculogenesis. [114] [116] Mice that are null for the VEGF-A gene die at 9.5 days post coitum (d.p.c.) due to a failure of vasculogenesis while mice lacking a single VEGF-A allele (i.e., they are heterozygotes for the VEGF-A gene) die at 11.5 d.p.c. also from vascular defects.[116] These data demonstrate gene dosage sensitivity to VEGF-A during development. In the developing kidney, podocytes and renal tubular epithelial cells express VEGF-A[117] and continue to express it constitutively in the adult kidney, while the cognate tyrosine-kinase receptors for VEGF-A, VEGFR-1 (Flt1), and VEGFR-2 (Flk1/KDR) are predominantly expressed by all endothelial cells. Which non-endothelial cells might also express the VEGF receptors in the kidney in vivo is still debated, although renal cell lines clearly do and metanephric mesenchymal cells express VEGFR-2 in organ culture as outlined earlier.
Conditional gene targeting experiments and cell-selective deletion of VEGF-A from podocytes demonstrates that VEGF-A signaling is required for formation and maintenance of the glomerular filtration barrier. [118] [119] Glomerular endothelial cells express VEGFR-2 as they migrate into the vascular cleft. Although a few endothelia migrate into the developing glomeruli of VEGFlox/lox/Pod-Cre mice (mice with selective deletion of VEGF from podocytes), likely due to a small amount of VEGF-A produced by presumptive podocytes at the S-shaped stage of glomerular development prior to Cre-mediated genetic deletion, the endothelia failed to develop fenestrations and rapidly disappeared leaving capillary “ghosts” ( Fig. 1-10 ). Similar to the dosage sensitivity observed in the whole embryo, deletion of a single VEGF-A allele from podocytes also led to glomerular endothelial defects known as endotheliosis that progressed to end-stage kidney failure at 3 months of age; as the dose of VEGF-A decreased, the associated endothelial phenotypes became more severe ( Fig. 1-11 ). Upregulation of the major 164 angiogenic VEGF-A isoform in developing podocytes of transgenic mice led to massive proteinuria and collapse of the glomerular tuft by 5 days of age. Taken together, these results show a requirement for VEGF-A for development and maintenance of the specialized glomerular endothelia and demonstrate a major paracrine signaling function for VEGF-A in the glomerulus. Furthermore, tight regulation of the dose of VEGF-A is essential for proper formation of the glomerular capillary system; the molecular basis and mechanism of dosage sensitivity is unclear at present and is particularly intriguing given the documented inducible regulation of VEGF-A by hypoxia inducible factors (HIFs) at a transcriptional level. Despite this, it is clear that in vivo, a single VEGF-A allele is unable to compensate for loss of the other. Immortalized podocyte cell lines express a variety of VEGF receptors opening up the possibility that VEGF-A also plays an autocrine role in the developing glomerulus [120] [121] [122]; however, the functional relevance of these findings for the glomerulus in vivo is unclear at present.
|
|
|
|
|
|
|
FIGURE 1-10 Transmission electron micrographs of the glomerular filtration barrier from a wild-type (left) or transgenic mouse with selective knockout of VEGF from the podocytes. Podocytes (po) are seen in both but the endothelial layer (en) is entirely missing from the knockout mouse leaving a “capillary ghost.” Immunostaining for WT1 (podocytes/green) and PECAM (endothelial cells/red) confirms the absence of capillary wall in VEGF knockouts. (Adapted from Eremina V, Sood M, Haigh J, et al: Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 111:707–716, 2003.) |
|
|
|
|
|
|
|
|
FIGURE 1-11 Vascular endothelial growth factor dose and glomerular development: Photomicrographs of glomeruli from mice carrying different copy numbers of the VEGF gene within podocytes. A total knockout (loss of both alleles;-/-) results in failure of glomerular filtration barrier formation and perinatal death; a single hypomorphic allele (hypo/-) leads to massive mesangiolysis in the first weeks of life and death at 3 weeks of age; loss of one copy (+/-) results in endotheliosis (swelling of the endothelium) and death at 12 weeks of age. Overexpression (20-fold increase in VEGF;++++) results in collapsing glomerulopathy. (Adapted from Eremina V, Baeld HJ, Quaggin SE: Role of the VEGF-A signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol 106:32–37, 2007.) |
|
A second major receptor tyrosine kinase (RTK) signaling pathway required for maturation of developing blood vessels is the Angiopoietin-Tie signaling system. Angiopoietin 1 stabilizes newly formed blood vessels and is associated with loss of vessel plasticity and concurrent recruitment of pericytes or vascular support cells to the vascular wall.[123] The molecular switch or pathway leading to vessel maturation through activation of Tie2 (previously known as Tek), the major receptor for Ang1 is not known and appears to be independent of the PDGF signaling system that is also required for pericyte recruitment. Ang1 knockout mice die at 12.5 d.p.c precluding analysis of its role in glomerular development. In contrast, it is proposed that Ang2 functions as a natural antagonist of the Tie2 receptor, as Ang2 can bind to this receptor but fails to phosphorylate it in endothelial cultures. [124] [125] Consistent with this hypothesis is the fact that overexpression of Ang2 in transgenic mice results in a phenotype similar to the Ang1 or Tie2 KO mice. Ang1, Ang2, Tie2, and Tie1 (an orphan receptor for this system) are all expressed in the developing kidney. [126] [127] [128] [129] [130] Whereas Ang1 is quite broadly expressed in condensing mesenchyme, podocytes, and tubular epithelial cells, Ang2 is more restricted to pericytes and smooth muscle cells surrounding cortical and large vessels as well as in the mesangium. Angiopoietin 2 KO mice are viable but exhibit defects in peritubular cortical capillary development; the mice died prior to differentiation of vasa rectae precluding analysis of the role of Ang2 in these other capillary beds.[131] Both angiopoietin ligands function in concert with VEGF, although precise degree of crosstalk between these pathways is still under investigation. (VEGF and Ang1 work together to promote sprouting.) Chimeric studies showed that the orphan receptor Tie1 is required for development of the glomerular capillary system, because Tie1 null cells are not able to contribute to glomerular capillary endothelium.[132]
The Ephrin-Eph family is a third tyrosine kinase dependent growth factor signaling system that is expressed in the developing kidney; in the whole embryo, it is involved in neural sprouting and axon finding as well as in arterial/venous specification of arterial and venous components of the vasculature. [133] [134] Ephrins and their cognate receptors are expressed widely during renal development. Overexpression of EPH4 leads to defects in glomerular arteriolar formation whereas conditional deletion of EphrinB2 from perivascular smooth muscle cells and mesangial cells leads to glomerular vascular abnormalities. [135] [136] How this occurs is not entirely clear as Ephrin B2 has a dynamic pattern of expression in the developing glomerulus, beginning in podocyte precursors and rapidly switching to glomerular endothelial cells and mesangial cells.[115]
An additional pathway that is likely to play a role in glomerular endothelial development and perhaps of the entire vasculature of the kidney, is the CXCR4-SDF1 axis. CXCR4, a chemokine receptor is expressed by bone marrow derived cells, but is also expressed in endothelial cells. SDF-1 (Cxcl12), the only known ligand for CXCR4 is expressed in a dynamic segmental pattern in the podocyte-endothelial compartment and later in the mesangial cells of the glomerulus.[137] Embryonic deletion of CXCR4 results in glomerular developmental defects with a dilated capillary network (presented at American Society of Nephrology, 2005).
Mice carrying a hypomorphic allele of Notch 2, which is missing 2 epidermal growth factor (EGF) motifs are born with a reduced number of glomeruli that lack both endothelial and mesangial cells as discussed in the section on nephron segmentation. [103] [104]
There is evidence from other model systems that vascu-lar development is required for patterning and terminal differentiation of adjacent tissues. For example, vascular signals and basement membrane produced by adjacent endothe-lial cells are required for differentiation of the islet cells of the pancreas. [138] [139] In the kidney, it is possible that vascu-lar signals are required for branching morphogenesis, and patterning of the nephron and may explain some of the defects observed in KO mice such as Pod1 (Tcf21) mutants. Given the complex reciprocal interactions between tissue types, these signals will be a challenge to sort out, but with the increasing arsenal of genetic tools, should be possible.
The Juxta-Glomerular Apparatus and the Renin-Angiotensin System
The juxtaglomerular apparatus consists of juxtaglomerular cells that line the afferent arteriole, the macula densa cells of the distal tubule, and the extraglomerular mesangial cells that are in contact with intraglomerular mesangium.[140] Renin-expressing cells may be seen in arterioles in early mesonephric kidneys in 5-week human fetus and in metanephric kidney by week 8, at a stage prior to hemodynamic flow changes within the kidney. Recently, Gomez and colleagues generated a Renin-knockin mouse that expresses Cre recombinase in the renin locus.[141] Offspring of matings between the renin-Cre and a reporter strain that expresses beta-galactosidase or GFP upon Cre-mediated DNA excision showed that renin-expressing cells may be found within MM and give rise not only to JG cells, but also to mesangial cells, epithelial cells, and extrarenal cells including interstitial Leydig cells of the XY gonad and cells within the adrenal gland. Although most of these cells cease to express renin in the adult, they appear to re-express renin in stress conditions and are recruited to the afferent arteriole.
The only known substrate for renin, angiotensinogen, is converted to angiotensin I and angiotensin II by angiotensin converting enzyme.[142] The renin-angiotensin-aldosterone axis is required for normal renal development. In humans, the use of angiotensin-converting enzyme inhibitors in early or late pregnancy have been associated with congenital defects including renal anomalies. [143] [144] Two subtypes of angiotensin receptors exist[145]: AT1 receptors are responsible for most of the classically recognized functions of the RAS including pressor effects and aldosterone release mediated through angiotensin; functions of the type 2 receptors have been more difficult to characterize, but generally seem to oppose the actions of the AT1 receptors. Genetic deletion of angiotensinogen [146] [147] or the ACE [148] [149] result in hypotension, and defects in formation of the renal papilla and pelvis. Humans have one AT1 gene while mice have two: AT1α and AT1b. Mice carrying a knockout for either AT1 receptor alone exhibit no major defects, [150] [151] while combined deficiency phenocopies the angiotensinogen and ACE phenotypes. [152] [153] While AT2 receptor expression is markedly up-regulated in the embryonic kidney, genetic deletion of the AT2 receptor does not cause major impairment of renal development. [154] [155] However, an association between AT2 receptor-deficiency and malformations of the collecting system, including VUR (vesicoureteral reflux) and ureteropelvic junction obstruction, has been reported.[156]
Nephron Development and Glomerulogenesis
Mesangial Cell Ingrowth
Mesangial cells grow into the developing glomerulus and come to sit between the capillary loops. Gene deletion studies have demonstrated a critical role for PDGF-B/beta receptor signaling in this process. Deletion of PDGF-B, which is expressed by glomerular endothelia, or the PDGF beta receptor that is expressed by mesangial cells, results in glomeruli with a single balloon-like capillary loop, instead of the intricately folded glomerular endothelial capillaries of wild-type kidneys. Furthermore, the glomeruli contain no mesangial cells.[157] Endothelial-cell specific deletion of PDGF-B results in the same glomerular phenotype and show that production of PDGF-B by the endothelium is required for mesangial migration.[158] In turn, mesangial cells and the matrix they produce are required to pattern the glomerular capillary system. Loss of podocyte-derived factors such as VEGF-A also lead to failure of mesangial cell ingrowth, likely through primary loss of endothelial cells and failure of PDGF-B signaling.
A number of other knockouts demonstrate defects in both vascular/capillary development and mesangial cell ingrowth. Transcription factors expressed by podocytes including Pod1 and Foxc2 show defects in mesangial cell migration ( Fig. 1-12 ). [49] [111] What factors are disrupted in these mutant mice to result in the phenotype are not yet known but emphasize the importance of “crosstalk” between cell compartments within the glomerulus.
|
|
|
|
|
|
|
FIGURE 1-12 Glomeruli from wild-type (A) or Pod1 knockout mice (B). Note dilated capillary loop and poor ingrowth of mesangial cells (me). |
|
Glomerular Epithelial Development
Presumptive podocytes are observed at the most proximal end of the S-shape body at the furthest point away from the ureteric bud tip. They form as columnar epithelial cells in apposition to the developing vascular cleft. A number of transcription factors have been identified that are expressed within developing and mature podocytes including: Wilms tumor suppressor 1, Pod1 (tcf21), Kreisler (maf1), Foxc2, and lmx1b ( Fig. 1-13 ). [6] [49] [110] [111] [159] [160]Genetic deletion studies have shown that Pod1, Foxc2, lmx1b, and Kreisler are all required for elaboration of podocyte foot processes and spreading of podocytes around the glomerular capillary beds. Pod1 appears to function upstream of Kreisler, as the latter factor is down-regulated in glomeruli from Pod1 KO mice.[159] Transcriptional programs regulated by these factors are incompletely known, however, Pod1, Foxc2, and lmx1b knockout mice all display remarkably similar glomerular phenotypes with major podocyte developmental/maturation defects together with capillary loop, glomerular basement membrane, and mesangial ingrowth abnormalities. It is believed that loss of podocyte-expressed factors regulated by these proteins leads to the dramatic arrest in development resulting in abnormal capillary loop stage glomeruli. Immunostaining and gene expression profiling performed in glomeruli from each of these mutant mice have identified reduced expression of some common downstream effector molecules including collagen alpha 4. In turn, a podocyte-specific protein, podocin (NPHS2) is reduced in the glomeruli of Foxc2, lmx1b, and Pod1 KO mice. [49] [161] Mutations in lmx1b are associated with nail patellar syndrome in humans, a disease characterized by absent patellae and nephrotic syndrome in a subset of patients. All of these genes are expressed from the S-shape stage onward and remain constitutively expressed in adult glomeruli.
|
|
|
|
|
|
|
FIGURE 1-13 Molecular basis of glomerular development. Key factors are shown; time point of major effect observed in knockout or transgenic studies is identified. Many factors play roles at more than one time point. *Mutations identified in patients with glomerular disease. (Top panel adapted from Saxen.) |
|
WT1 is also expressed in presumptive and mature podocytes. As WT1 knockout mice fail to develop any kidneys, the role of WT1 in the developing and mature podocyte is not entirely clear. However, a series of experimental models support an important role for WT1 in the podocyte. The null phenotype was largely rescued using a yeast artificial chromosome (YAC) containing the human WT1 gene[5]; depending on the level of WT1 expression, these mice developed crescentic glomerulonephritis or mesangial sclerosis, defects in the glomerulus reminiscent of some of the human phenotypes observed with Denys Drash syndrome resulting from mutations in the KTS isoform of WT1.[5] Transgenic mice express-ing a Denys-Drash mutant WT1 allele under the regulation of a podocyte-specific promoter also developed glom-erular disease with abnormalities observed in the adjacent endothelium.[4]
Alpha 3 integrin KO mice also demonstrate defects in glomerular development with specific abnormalities in podocyte maturation.[98] Alpha 3 integrin forms a heterodimer with b1 integrin and is expressed by podocytes. Loss of this integrin results in poorly formed foot processes and abnormalities in adjacent glomerular basement membrane. Elaboration of foot processes during development requires the interaction of an adaptor signaling molecule, NCK, with slit diaphragm proteins. The slit diaphragm is a specialized intercellular junction that connects foot processes of adjacent podocytes. In mature glomeruli, the slit diaphragm appears as a dense band on transmission electron micrographs. In 1998, Karl Tryggvason's group identified the nephrin (NPHS1) gene and showed that mutations in this gene cause congenital nephrosis of the Finnish (CNF) variety.[162] Glomeruli from infants with CNF, lack slit diaphragms and die from renal failure and nephrotic syndrome unless they receive renal replacement therapy. Nephrin is a member of the immunoglobulin superfamily and makes up a major structural component of the slit diaphragm. Recently, it was shown that the nephrin molecule contains 3 intracellular tyrosine residues (1176, 1193, 1217) that can be phosphorylated leading to recruitment of the SH2 adaptor proteins, NCK 1 and 2. [17] [18] In vitro, this association leads to reorganization of the actin cytoskeleton, the backbone of foot process structure. Mice born with podocyte-selective deletion of the NCK1 and 2 genes exhibit congenital nephrosis and fail to form any foot processes ( Fig. 1-14 ), emphasizing the biologic link between the slit diaphragm and cytoskeleton in vivo. The phenotype observed in Nck-deficient mice is similar to that of FAT1 KO mice, with complete absence of foot process formation and podocyte effacement. FAT1 is a large protocadherin expressed in podocytes. In contrast, Nephrin KO mice exhibit narrowed slits with loss of the diaphragm but the degree of foot process effacement or fusion varies and may be dependent on mouse strain. Additional proteins of the slit diaphragm and cytoskeleton have been identified that play major roles in glomerulogenesis and in human disease. Three Nephrin-like molecules exist (neph1–3) that are also expressed in podocytes and interact with other slit diaphragm proteins. [163] [164] Mice generated through a gene-trap screen with loss of Neph1 function develop focal segmental glomerulo sclerosis (FSGS), suggesting that these molecules are also important for function of the slit diaphragm.[48] NPHS2 (podocin) is a homologue of the Caenorhabditis elegans mechanosensory channel (MEC), MEC2, and co-localizes with nephrin at the slit diaphragm. Mutations in NPHS2 have been identified in patients with steroid-resistant congenital autosomal recessive FSGS.[165] Although foot process effacement is a feature of the NPHS2 KO mice, vascular defects are also observed and suggest that podocin or the slit diaphragm (or both) may regulate components of the crosstalk between podocytes and endothelium.[166] Mutations in alpha actinin 4, a component of the actin cytoskeleton, were identified in autosomal dominant FSGS, a disease with its onset in adulthood.[167] Pollak and colleagues hypothesized that the mutant actinin functions in a dominant negative manner to cause the phenotype. Interestingly, ACTN4 KO mice also develop glomerular disease, despite the proteins rather ubiquitous expression.[168] Clearly, elucidation of the mechanism(s) is important, and will provide exciting insights into glomerular biology.
|
|
|
|
|
|
|
FIGURE 1-14 Foot processes (fp) are seen surrounding a capillary loop of a newborn wild-type mouse. In the right panel, a capillary loop from a NCK1/2 knockout mouse shows complete lack of foot process formation. En, glomerular endothelial cell. (Adapted from Jones N, Blasutig IM, Eremina V, et al: Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440:818–823, 2006.) |
|
CD2AP (CD2-associated protein), is an SH3 domain containing protein in lymphoid cells and podocytes that interacts with the cytoplasmic tail of nephrin and with the actin cytoskeleton.[169] Null CD2AP mice rapidly develop massive proteinuria and mesangial sclerosis, a finding that highlights the interplay of all of these molecules in the glomerulus. CD2AP heterozygous mice are susceptible to glomerular disease and exhibit glomerular lesions at 12 to 18 months that are similar to immunotactoid glomerulopathy in humans. [44] [170] CD2AP has been implicated in endocytosis and lysosomal sorting and may be required to clear immunoglobulins that are normally filtered at the glomerulus. Consistent with this hypothesis, the investigators showed that a large proportion of CD2AP heterozygous mice develop FSGS-like lesions when injected with a nephrotoxic antibody.[170] Mutations in TRPC6 channel underlie another inherited form of AD-FSGS in patients. Some, but not all, of the identified mutations lead to activation of the channel and increased influx of calcium inside the cell. [171] [172] TRPC6 is expressed in podocytes but also other glomerular cell types. Elucidation of the mechanism whereby TRPC6 leads to glomerular disease is exciting and may provide a mechanism that is amenable to pharmacologic intervention.
From all of these studies, it follows that intrinsic proteins and functions of podocytes play a key role in the development and maintenance of the permselective properties of the glomerular filtration barrier; however, as outlined earlier in the section on vascular development, podocytes also function as vasculature supporting cells producing VEGF and other angiogenic growth factors. It is likely that endothelial cells also produce factors required for terminal differentiation of podocytes, although these factors are currently unknown.
Maturation of Glomerular Endothelial Cells and Glomerular Basement Membrane
Following migration into the glomerular vascular cleft, endothelial cells are rounded and capillaries do not possess a lumen. During glomerulogenesis, lumens form through apoptosis of a subset of endothelial cells and the endothe-lial cells flatten considerably and develop fenestrations and a complex glycocalyx. This process is believed to be dependent on a TGFb1-dependent signal.[227] Loss of these specialized features of the glomerular endothelium as in endotheliosis, leads to disruption of the filtration barrier and protein loss, emphasizing that this layer of the GFB plays a major role in permselectivity.
Another chapter in the book deals with properties and development of the glomerular basement membrane although informative knockouts are included in Table 1-1 for completeness. It is important to note that components are produced by both podocytes and glomerular endothelium and that a number of vital growth factors, such as VEGF-A are stored and processed in the GBM.
References
1. Zandi-Nejad K, Luyckx VA, Brenner BM: Adult hypertension and kidney disease: The role of fetal programming. Hypertension 2006; 47:502-508.
2. Luyckx VA, Brenner BM: Low birth weight, nephron number, and kidney disease. Kidney Int 2005;S68-S77.
3. Rossing P, Tarnow L, Nielsen FS, et al: Low birth weight. A risk factor for development of diabetic nephropathy?. Diabetes 1995; 44:1405-1407.
4. Natoli TA, Liu J, Eremina V, et al: A mutant form of the Wilms' tumor suppressor gene WT1 observed in Denys-Drash Syndrome interferes with glomerular capillary development. J Am Soc Nephrol 2002; 13:2058-2067.
5. Moore AW, McInnes L, Kreidberg J, et al: YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 1999; 126:1845-1857.
6. Kreidberg JA, Sariola H, Loring JM, et al: WT-1 is required for early kidney development. Cell 1993; 74:679-691.
7. Saxen L: Organogenesis of the Kidney, Cambridge, Cambridge University Press, 1987.
8. Rothenpieler UW, Dressler GR: Pax-2 is required for mesenchyme-to-epithelium conversion during kidney development. Development 1993; 119:711-720.
9. Torres M, Gomez-Pardo E, Dressler GR, Gruss P: Pax-2 controls multiple steps of urogenital development. Development 1995; 121:4057-4065.
10. Capel B, Albrecht KH, Washburn LL, Eicher EM: Migration of mesonephric cells into the mammalian gonad depends on Sry. Mech Dev 1999; 84:127-131.
11. Cui S, Ross A, Stallings N, et al: Disrupted gonadogenesis and male-to-female sex reversal in Pod1 knockout mice. Development 2004; 131:4095-4105.
12. Mendelsohn C, Lohnes D, Decimo D, et al: Function of the retinoic acid receptors (RARs) during development (II. Multiple abnormalities at various stages of organogenesis in RAR double mutants). Development 1994; 120:2749-2771.
13. Kreidberg JA: Podocyte differentiation and glomerulogenesis. J Am Soc Nephrol 2003; 14:806-814.
14. Batourina E, Gim S, Bello N, et al: Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat Genet 2001; 27:74-78.
15. Reeves W, Caulfield JP, Farquhar MG: Differentiation of epithelial foot processes and filtration slits: Sequential appearance of occluding junctions, epithelial polyanion, and slit membranes in developing glomeruli. Lab Invest 1978; 39:90-100.
16. Batourina E, Tsai S, Lambert S, et al: Apoptosis induced by vitamin A signaling is crucial for connecting the ureters to the bladder. Nat Genet 2005; 37:1082-1089.
17. Jones N, Blasutig IM, Eremina V, et al: Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 2006; 440:818-823.
18. Verma R, Kovari I, Soofi A, et al: Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest 2006; 116:1346-1359.
19. Tryggvason K, Pikkarainen T, Patrakka J: Nck links nephrin to actin in kidney podocytes. Cell 2006; 125:221-224.
20. Fierlbeck W, Liu A, Coyle R, Ballermann BJ: Endothelial cell apoptosis during glomerular capillary lumen formation in vivo. J Am Soc Nephrol 2003; 14:1349-1354.
21. Rastaldi MP, Armelloni S, Berra S, et al: Glomerular podocytes possess the synaptic vesicle molecule Rab3A and its specific effector rabphilin-3α. Am J Pathol 2003; 163:889-899.
22. Kobayashi N, Gao SY, Chen J, et al: Process formation of the renal glomerular podocyte: Is there common molecular machinery for processes of podocytes and neurons?. Anat Sci Int 2004; 79:1-10.
23. Baum M, Quigley R, Satlin L: Maturational changes in renal tubular transport. Curr Opin Nephrol Hypertens 2003; 12:521-526.
24. Osathanondh V, Potter EL: Development of human kidney as shown by microdissection. iii. Formation and interrelationship of collecting tubules and nephrons. Arch Pathol 1963; 76:290-302.
25. Bard J: A new role for the stromal cells in kidney development. Bioessays 1996; 18:705-707.
26. Hatini V, Huh SO, Herzlinger D, et al: Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev 1996; 10:1467-1478.
27. Levinson R, Mendelsohn C: Stromal progenitors are important for patterning epithelial and mesenchymal cell types in the embryonic kidney. Semin Cell Dev Biol 2003; 14:225-231.
28. Robert B, St. John PL, Hyink DP, Abrahamson DR: Evidence that embryonic kidney cells expressing flk-1 are intrinsic, vasculogenic angioblasts. Am J Physiol 1996; 271:F744-F753.
29. Grobstein C: Inductive epitheliomesenchymal interaction in cultured organ rudiments of the mouse. Science 1953; 118:52-55.
30. Miyamoto N, Yoshida M, Kuratani S, et al: Defects of urogenital development in mice lacking Emx2. Development 1997; 124:1653-1664.
31. Sariola H, Saarma M, Sainio K, et al: Dependence of kidney morphogenesis on the expression of nerve growth factor receptor. Science 1991; 254:571-573.
32. Durbeej M, Soderstrom S, Ebendal T, et al: Differential expression of neurotrophin receptors during renal development. Development 1993; 119:977-989.
33. Sainio K, Saarma M, Nonclercq D, et al: Antisense inhibition of low-affinity nerve growth factor receptor in kidney cultures: Power and pitfalls. Cell Mol Neurobiol 1994; 14:439-457.
34. Davies JA, Ladomery M, Hohenstein P, et al: Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum Mol Genet 2004; 13:235-246.
35. Gao X, Chen X, Taglienti M, et al: Angioblast-mesenchyme induction of early kidney development is mediated by Wt1 and Vegfa. Development 2005; 132:5437-5449.
36. Vainio S, Lin Y: Coordinating early kidney development: Lessons from gene targeting. Nat Rev Genet 2002; 3:533-543.
37. Dressler GR: Kidney development branches out. Dev Genet 1999; 24:189-193.
38. Gawlik A, Quaggin SE: Conditional gene targeting in the kidney. Curr Mol Med 2005; 5:527-536.
39. Gawlik A, Quaggin SE: Deciphering the renal code; advances in conditional gene targeting in the kidney. Physiology (Bethesda) 2004; 19:245-252.
40. Justice M: Capitalizing on large-scale mouse mutagenesis screens. Nat Rev Genet 2000; 2:109-115.
41. Justice M, Carpenter D, Favor J, et al: Effects of ENU dosage on mouse strains. Mamm Genome 2000; 11:484-488.
42. Hrabe de Angelis MH, Flaswinkel H, Fuchs H, et al: Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet 2000; 25:444-447.
43. Nolan PM, Peters J, Strivens M, et al: A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet 2000; 25:440-443.
44. Huber TB, Kwoh C, Wu H, et al: Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J Clin Invest 2006; 116:1337-1345.
45. Beier DR, Herron BJ: Genetic mapping and ENU mutagenesis. Genetica 2004; 122:65-69.
46. Cordes SP: N-ethyl-N-nitrosourea mutagenesis: Boarding the mouse mutant express. Microbiol Mol Biol Rev 2005; 69:426-439.
47. Stanford WL, Cohn JB, Cordes SP: Gene-trap mutagenesis: past, present and beyond. Nat Rev Genet 2001; 2:756-768.
48. Donoviel DB, Freed DD, Vogel H, et al: Proteinuria and parinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol 2001; 21:4829-4836.
49. Takemoto M, He L, Norlin J, et al: Large-scale identification of genes implicated in kidney glomerulus development and function. EMBO J 2006; 25:1160-1174.
50. Shen K, Fetter RD, Bargmann CI: Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell 2004; 116:868-881.
51. Nelson FK, Albert PS, Riddle DL: Fine structure of the Caenorhabditis elegans secretory-excretory system. J Ultrastruct Res 1983; 82:156-171.
52. Barr MM: Caenorhabditis elegans as a model to study renal development and disease: Sexy cilia. J Am Soc Nephrol 2005; 16:305-312.
53. Barr MM, DeModena J, Braun D, et al: The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr Biol 2001; 11:1341-1346.
54. Simon JM, Sternberg PW: Evidence of a mate-finding cue in the hermaphrodite nematode Caenorhabditis elegans. Proc Natl Acad Sci U S A 2002; 99:1598-1603.
55. Jung AC, Denholm B, Skaer H, Affolter M: Renal tubule development in Drosophila: A closer look at the cellular level. J Am Soc Nephrol 2005; 16:322-328.
56. Dworak HA, Charles MA, Pellerano LB, Sink H: Characterization of Drosophila hibris, a gene related to human nephrin. Development 2001; 128:4265-4276.
57. Schneider T, Reiter C, Eule E, et al: Restricted expression of the irreC-rst protein is required for normal axonal projections of columnar visual neurons. Neuron 1995; 15:259-271.
58. Venugopala Reddy G, Reiter C, Shanbhag S, et al: Irregular chiasm-C-roughest, a member of the immunoglobulin superfamily, affects sense organ spacing on the Drosophila antenna by influencing the positioning of founder cells on the disc ectoderm. Dev Genes Evol 1999; 209:581-591.
59. Majumdar A, Drummond IA: Podocyte differentiation in the absence of endothelial cells as revealed in the zebrafish avascular mutant, cloche. Dev Genet 1999; 24:220-229.
60. Drummond IA: Kidney development and disease in the zebrafish. J Am Soc Nephrol 2005; 16:299-304.
61. Jones EA: Xenopus: a prince among models for pronephric kidney development. J Am Soc Nephrol 2005; 16:313-321.
62. Xu PX, Adams J, Peters H, et al: Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet 1999; 23:113-117.
63. Laclef C, Souil E, Demignon J, Maire P: Thymus, kidney and craniofacial abnormalities in Six 1 deficient mice. Mech Dev 2003; 120:669-679.
64. Xu PX, Zheng W, Huang L, et al: Six1 is required for the early organogenesis of mammalian kidney. Development 2003; 130:3085-3094.
65. Nishinakamura R, Matsumoto Y, Nakao K, et al: Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development 2001; 128:3105-3115.
66. Shawlot W, Behringer RR: Requirement for Lim1 in head-organizer function. Nature 1995; 374:425-430.
67. Moore MW, Klein RD, Farinas I, et al: Renal and neuronal abnormalities in mice lacking GDNF. Nature 1996; 382:76-79.
68. Pichel JG, Shen L, Sheng HZ, et al: Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 1996; 382:73-76.
69. Sanchez MP, Silos-Santiago I, Frisen J, et al: Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 1996; 382:70-73.
70. Esquela AF, Lee SJ: Regulation of metanephric kidney development by growth/differentiation factor 11. Dev Biol 2003; 257:356-370.
71. Michos O, Panman L, Vintersten K, et al: Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development 2004; 131:3401-3410.
72. Schuchardt A, D'Agati V, Larsson-Blomberg L, et al: RET-deficient mice: An animal model for Hirschsprung's disease and renal agenesis. J Intern Med 1995; 238:327-332.
73. Cacalano G, Farinas I, Wang LC, et al: GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron 1998; 21:53-62.
74. Buller C, Xu X, Marquis V, et al: Molecular effects of Eya1 domain mutations causing organ defects in BOR syndrome. Hum Mol Genet 2001; 10:2775-2781.
75. Ikeda K, Watanabe Y, Ohto H, Kawakami K: Molecular interaction and synergistic activation of a promoter by Six, Eya, and Dach proteins mediated through CREB binding protein. Mol Cell Biol 2002; 22:6759-6766.
76. Li X, Oghi KA, Zhang J, et al: Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003; 426:247-254.
77. Fougerousse F, Durand M, Lopez S, et al: Six and Eya expression during human somitogenesis and MyoD gene family activation. J Muscle Res Cell Motil 2002; 23:255-264.
78. Pandur PD, Moody SA: Xenopus Six1 gene is expressed in neurogenic cranial placodes and maintained in the differentiating lateral lines. Mech Dev 2000; 96:253-257.
79. Sajithlal G, Zou D, Silvius D, Xu PX: Eya 1 acts as a critical regulator for specifying the metanephric mesenchyme. Dev Biol 2005; 284:323-336.
80. Brophy PD, Ostrom L, Lang KM, Dressler GR: Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development 2001; 128:4747-4756.
81. Poladia DP, Kish K, Kutay B, et al: Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev Biol 2006; 291:325-339.
82. Grieshammer U, Cebrian C, Ilagan R, et al: FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 2005; 132:3847-3857.
83. Perantoni AO, Timofeeva O, Naillat F, et al: Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 2005; 132:3859-3871.
84. Stark K, Vainio S, Vassileva G, McMahon AP: Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 1994; 372:679-683.
85. Luo G, Hofmann C, Bronckers AL, et al: BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev 1995; 9:2808-2820.
86. Dudley AT, Lyons KM, Robertson EJ: A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev 1995; 9:2795-2807.
87. Muller U, Wang D, Denda S, et al: Integrin alpha8beta1 is critically important for epithelial-mesenchymal interactions during kidney morphogenesis. Cell 1997; 88:603-613.
88. Brandenberger R, Schmidt A, Linton J, et al: Identification and characterization of a novel extracellular matrix protein nephronectin that is associated with integrin alpha8beta1 in the embryonic kidney. J Cell Biol 2001; 154:447-458.
89. Miyazaki Y, Oshima K, Fogo A, et al: Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J Clin Invest 2000; 105:863-873.
90. Chiang C, Litingtung Y, Lee E, et al: Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996; 383:407-413.
91. Qiao J, Uzzo R, Obara-Ishihara T, et al: FGF-7 modulates ureteric bud growth and nephron number in the developing kidney. Development 1999; 126:547-554.
92. Zhao H, Kegg H, Grady S, et al: Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 2004; 276:403-415.
93. Gupta IR, Macias-Silva M, Kim S, et al: BMP-2/ALK3 and HGF signal in parallel to regulate renal collecting duct morphogenesis. J Cell Sci 2000; 113(Pt 2):269-278.
94. Piscione TD, Yager TD, Gupta IR, et al: BMP-2 and OP-1 exert direct and opposite effects on renal branching morphogenesis. Am J Physiol 1997; 273:F961-F975.
95. Hu MC, Piscione TD, Rosenblum ND: Elevated SMAD1/beta-catenin molecular complexes and renal medullary cystic dysplasia in ALK3 transgenic mice. Development 2003; 130:2753-2766.
96. Hu MC, Rosenblum ND: Smad1, beta-catenin and Tcf4 associate in a molecular complex with the Myc promoter in dysplastic renal tissue and cooperate to control Myc transcription. Development 2005; 132:215-225.
97. Hu MC, Mo R, Bhella S, et al: GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development 2006; 133:569-578.
98. Kreidberg JA, Donovan MJ, Goldstein SL, et al: Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development 1996; 122:3537-3547.
99. Korhonen M, Ylanne J, Laitinen L, Virtanen I: The alpha 1-alpha 6 subunits of integrins are characteristically expressed in distinct segments of developing and adult human nephron. J Cell Biol 1990; 111:1245-1254.
100. Chattopadhyay N, Wang Z, Ashman LK, et al: alpha3beta1 integrin-CD151, a component of the cadherin-catenin complex, regulates PTPmu expression and cell-cell adhesion. J Cell Biol 2003; 163:1351-1362.
101. Kume T, Deng K, Hogan BL: Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development 2000; 127:1387-1395.
102. Grieshammer U, Le M, Plump AS, et al: SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell 2004; 6:709-717.
103. McCright B: Notch signaling in kidney development. Curr Opin Nephrol Hypertens 2003; 12:5-10.
104. McCright B, Gao X, Shen L, et al: Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 2001; 128:491-502.
105. McCright B, Lozier J, Gridley T: A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002; 129:1075-1082.
106. Cheng HT, Kopan R: The role of Notch signaling in specification of podocyte and proximal tubules within the developing mouse kidney. Kidney Int 2005; 68:1951-1952.
107. Cheng HT, Miner JH, Lin M, et al: Gamma-secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development 2003; 130:5031-5042.
108. Wang P, Pereira FA, Beasley D, Zheng H: Presenilins are required for the formation of comma- and S-shaped bodies during nephrogenesis. Development 2003; 130:5019-5029.
109. Blomqvist SR, Vidarsson H, Fitzgerald S, et al: Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest 2004; 113:1560-1570.
110. Quaggin SE, Vanden Heuvel GB, Igarashi P: Pod-1, a mesoderm-specific basic-helix-loop-helix protein expressed in mesenchymal and glomerular epithelial cells in the developing kidney. Mech Dev 1998; 71:37-48.
111. Quaggin SE, Schwartz L, Post M, Rossant J: The basic-helix-loop-helix protein Pod-1 is critically important for kidney and lung organogenesis. Development 1999; 126:5771-5783.
112. Cui S, Schwartz L, Quaggin SE: Pod1 is required in stromal cells for glomerulogenesis. Dev Dyn 2003; 226:512-522.
113. Mendelsohn C, Batourina E, Fung S, et al: Stromal cells mediate retinoid-dependent functions essential for renal development. Development 1999; 126:1139-1148.
114. Shalaby F, Rossant J, Yamaguchi TP, et al: Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995; 376:62-66.
115. Takahashi T, Takahashi K, Gerety S, et al: Temporally compartmentalized expression of ephrin-B2 during renal glomerular development. J Am Soc Nephrol 2001; 12:2673-2682.
116. Carmeliet P, Ferreira V, Breier G, et al: Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996; 380:435-439.
117. Villegas G, Lange-Sperandio B, Tufro A: Autocrine and paracrine functions of vascular endothelial growth factor (VEGF) in renal tubular epithelial cells. Kidney Int 2005; 67:449-457.
118. Eremina V, Sood M, Haigh J, et al: Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 2003; 111:707-716.
119. Eremina V, Cui S, Gerber H, et al: VEGF-A signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol 2006; 17:724-735.
120. Foster RR, Hole R, Anderson K, et al: Functional evidence that vascular endothelial growth factor may act as an autocrine factor on human podocytes. Am J Physiol Renal Physiol 2003; 284:F1263-F1273.
121. Foster RR, Saleem MA, Mathieson PW, et al: Vascular endothelial growth factor and nephrin interact and reduce apoptosis in human podocytes. Am J Physiol Renal Physiol 2005; 288:F48-F57.
122. Guan F, Villegas G, Teichman J, et al: Autocrine VEGF-A system in podocytes regulates podocin and its interaction with CD2AP. Am J Physiol Renal Physiol 2006; 291:F422-F428.
123. Suri C, Jones PF, Patan S, et al: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis [see comments]. Cell 1996; 87:1171-1180.
124. Maisonpierre PC, Suri C, Jones PF, et al: Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis [see comments]. Science 1997; 277:55-60.
125. Augustin HG, Breier G: Angiogenesis: Molecular mechanisms and functional interactions. Thromb Haemost 2003; 89:190-197.
126. Yuan HT, Suri C, Yancopoulos GD, Woolf AS: Expression of angiopoietin-1, angiopoietin-2, and the Tie-2 receptor tyrosine kinase during mouse kidney maturation. J Am Soc Nephrol 1999; 10:1722-1736.
127. Woolf AS, Yuan HT: Angiopoietin growth factors and Tie receptor tyrosine kinases in renal vascular development. Pediatr Nephrol 2001; 16:177-184.
128. Yuan HT, Suri C, Landon DN, et al: Angiopoietin-2 is a site-specific factor in differentiation of mouse renal vasculature. J Am Soc Nephrol 2000; 11:1055-1066.
129. Satchell SC, Harper SJ, Mathieson PW: Angiopoietin-1 is normally expressed by periendothelial cells. Thromb Haemost 2001; 86:1597-1598.
130. Satchell SC, Harper SJ, Tooke JE, et al: Human podocytes express angiopoietin 1, a potential regulator of glomerular vascular endothelial growth factor. J Am Soc Nephrol 2002; 13:544-550.
131. Pitera JE, Woolf AS, Gale NW, et al: Dysmorphogenesis of kidney cortical peritubular capillaries in angiopoietin-2-deficient mice. Am J Pathol 2004; 165:1895-1906.
132. Partanen J, Puri MC, Schwartz L, et al: Cell autonomous functions of the receptor tyrosine kinase TIE in a late phase of angiogenic capillary growth and endothelial cell survival during murine development. Development 1996; 122:3013-3021.
133. Gerety SS, Anderson DJ: Cardiovascular ephrinB2 function is essential for embryonic angiogenesis. Development 2002; 129:1397-1410.
134. Wang HU, Anderson DJ: Eph family transmembrane ligands can mediate repulsive guidance of trunk neural crest migration and motor axon outgrowth. Neuron 1997; 18:383-396.
135. Andres AC, Munarini N, Djonov V, et al: EphB4 receptor tyrosine kinase transgenic mice develop glomerulopathies reminiscent of aglomerular vascular shunt. Mech Dev 2003; 120:511-516.
136. Foo SS, Turner CJ, Adams S, et al: Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 2006; 124:161-173.
137. Ding M, Cui S, Li C, et al: Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nature Medicine 2006; 12:1081-1087.
138. Nikolova G, Jabs N, Konstantinova I, et al: The vascular basement membrane: A niche for insulin gene expression and Beta cell proliferation. Dev Cell 2006; 10:397-405.
139. Lammert E, Cleaver O, Melton D: Induction of pancreatic differentiation by signals from blood vessels. Science 2001; 294:564-567.
140. Schnermann J, Homer W: Smith Award lecture. The juxtaglomerular apparatus: from anatomical peculiarity to physiological relevance. J Am Soc Nephrol 2003; 14:1681-1694.
141. Sequeira Lopez ML, Pentz ES, Nomasa T, et al: Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell 2004; 6:719-728.
142. Husain A, Graham R: Enzymes and Receptors of the Renin-Angiotensin System: Celebrating a Century of Discovery, Sidney, Harwood Academic, 2000.
143. Cooper WO, Hernandez-Diaz S, Arbogast PG, et al: Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med 2006; 354:2443-2451.
144. Friberg P, Sundelin B, Bohman SO, et al: Renin-angiotensin system in neonatal rats: induction of a renal abnormality in response to ACE inhibition or angiotensin II antagonism. Kidney Int 1994; 45:485-492.
145. Timmermans PB, Wong PC, Chiu AT, et al: Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev 1993; 45:205-251.
146. Kim HS, Krege JH, Kluckman KD, et al: Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci U S A 1995; 92:2735-2739.
147. Niimura F, Labosky PA, Kakuchi J, et al: Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest 1995; 96:2947-2954.
148. Krege JH, John SW, Langenbach LL, et al: Male-female differences in fertility and blood pressure in ACE-deficient mice. Nature 1995; 375:146-148.
149. Esther Jr CR, Howard TE, Marino EM, et al: Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab Invest 1996; 74:953-965.
150. Ito M, Oliverio MI, Mannon PJ, et al: Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci U S A 1995; 92:3521-3525.
151. Sugaya T, Nishimatsu S, Tanimoto K, et al: Angiotensin II type 1α receptor-deficient mice with hypotension and hyperreninemia. J Biol Chem 1995; 270:18719-18722.
152. Tsuchida S, Matsusaka T, Chen X, et al: Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J Clin Invest 1998; 101:755-760.
153. Oliverio MI, Kim HS, Ito M, et al: Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci U S A 1998; 95:15496-15501.
154. Ichiki T, Labosky PA, Shiota C, et al: Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature 1995; 377:748-750.
155. Hein L, Barsh GS, Pratt RE, et al: Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature 1995; 377:744-747.
156. Nishimura H, Yerkes E, Hohenfellner K, et al: Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol Cell 1999; 3:1-10.
157. Lindahl P, Hellstrom M, Kalen M, et al: Paracrine PDGF-B/PDGF-Rbeta signaling controls mesangial cell development in kidney glomeruli. Development 1998; 125:3313-3322.
158. Bjarnegard M, Enge M, Norlin J, et al: Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development 2004; 131:1847-1857.
159. Sadl V, Jin F, Yu J, et al: The mouse Kreisler (Krml1/MafB) segmentation gene is required for differentiation of glomerular visceral epithelial cells. Dev Biol 2002; 249:16-29.
160. Chen H, Lun Y, Ovchinnikov D, et al: Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet 1998; 19:51-55.
161. Cui S, Li C, Ema M, et al: Rapid isolation of glomeruli coupled with gene expression profiling identifies downstream targets in pod1 knockout mice. J Am Soc Nephrol 2005; 16:3247-3255.
162. Kestila M, Lenkkeri U, Mannikko M, et al: Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1998; 1:575-582.
163. Sellin L, Huber TB, Gerke P, et al: NEPH1 defines a novel family of podocin interacting proteins. FASEB J 2003; 17:115-117.
164. Huber TB, Hartleben B, Kim J, et al: Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol 2003; 23:4917-4928.
165. Lenkkeri U, Mannikko M, McCready P, et al: Structure of the gene for congenital nephrotic syndrome of the Finnish type (NPHS1) and characterization of mutations. Am J Hum Genet 1999; 64:51-61.
166. Roselli S, Heidet L, Sich M, et al: Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol Cell Biol 2004; 24:550-560.
167. Kaplan JM, Kim SH, North KN, et al: Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 2000; 24:251-256.
168. Kos CH, Le TC, Sinha S, et al: Mice deficient in alpha-actinin-4 have severe glomerular disease. J Clin Invest 2003; 111:1683-1690.
169. Shih NY, Li J, Karpitskii V, et al: Congenital nephrotic syndrome in mice lacking CD2-associated protein [see comments]. Science 1999; 286:312-315.
170. Kim JM, Wu H, Green G, et al: CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 2003; 300:1298-1300.
171. Winn MP, Conlon PJ, Lynn KL, et al: A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005; 308:1801-1804.
172. Reiser J, Polu KR, Moller CC, et al: TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 2005; 37:739-744.
173. Srinivas S, Wu Z, Chen C-M, et al: Dominant effects of RET receptor misexpression and ligand-independent RET signaling on ureteric bud development. Development 1999; 126:1375-1386.
174. Vega QC, Worby CA, Lechner MS, et al: Glial cell line-derived neurotrophic factor activates the receptor tyrosine kinase RET and promotes kidney morphogenesis. Proc Natl Acad Sci U S A 1996; 93:10657-10661.
175. Jing S, Wen D, Yu Y, et al: GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell 1996; 85:1113-1124.
176. Schuchardt A, D'Agati V, Larsson-Blomberg L, et al: Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994; 367:380-383.
177. Schuchardt A, D'Agati V, Pachnis V, Costantini F: Renal agenesis and hypodysplasia in ret-k-mutant mice result from defects in ureteric bud development. Development 1996; 122:1919-1929.
178. Sato A, Kishida S, Tanaka T, et al: Sall1, a causative gene for Townes-Brocks syndrome, enhances the canonical Wnt signaling by localizing to heterochromatin. Biochem Biophys Res Commun 2004; 319:103-113.
179. Kispert A, Vainio S, McMahon AP: Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 1998; 125:4225-4234.
180. Dreyer SD, Zhou G, Baldini A, et al: Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 1998; 19:47-50.
181. Lin F, Hiesberger T, Cordes K, et al: Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A 2003; 100:5286-5291.
182. Gresh L, Fischer E, Reimann A, et al: A transcriptional network in polycystic kidney disease. EMBO J 2004; 23:1657-1668.
183. Rankin EB, Tomaszewski JE, Haase VH: Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 2006; 66:2576-2583.
184. Lu W, Peissel B, Babakhanlou H, et al: Perinatal lethality with kidney and pancreas defects in mice with a targeted Pkd1 mutation. Nat Genet 1997; 17:179-181.
185. Miner JH, Sanes JR: Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): Implications for Alport syndrome. J Cell Biol 1996; 135:1403-1413.
186. Cosgrove D, Meehan DT, Grunkemeyer JA, et al: Collagen COL4A3 knockout: A mouse model for autosomal Alport syndrome. Genes Dev 1996; 10:2981-2992.
187. Lu W, Phillips CL, Killen PD, et al: Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model of Alport syndrome. Genomics 1999; 61:113-124.
188. Rheault MN, Kren SM, Thielen BK, et al: Mouse model of X-linked Alport syndrome. J Am Soc Nephrol 2004; 15:1466-1474.
189. Gould DB, Phalan FC, van Mil SE, et al: Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 2006; 354:1489-1496.
190. Noakes PG, Miner JH, Gautam M, et al: The renal glomerulus of mice lacking s-laminin/laminin beta 2: Nephrosis despite molecular compensation by laminin beta 1. Nat Genet 1995; 10:400-406.
191. Jarad G, Cunningham J, Shaw AS, Miner JH: Proteinuria precedes podocyte abnormalities inLamb2-/- mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest 2006; 116:2272-2279.
192. Miner JH, Li C: Defective glomerulogenesis in the absence of laminin alpha5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol 2000; 217:278-289.
193. Kikkawa Y, Virtanen I, Miner JH: Mesangial cells organize the glomerular capillaries by adhering to the G domain of laminin alpha5 in the glomerular basement membrane. J Cell Biol 2003; 161:187-196.
194. Kikkawa Y, Miner JH: Molecular dissection of laminin alpha5 in vivo reveals separable domain-specific roles in embryonic development and kidney function. Dev Biol 2006; 296:265-277.
195. Morita H, Yoshimura A, Inui K, et al: Heparan sulfate of perlecan is involved in glomerular filtration. J Am Soc Nephrol 2005; 16:1703-1710.
196. Lebel SP, Chen Y, Gingras D, et al: Morphofunctional studies of the glomerular wall in mice lacking entactin-1. J Histochem Cytochem 2003; 51:1467-1478.
197. Kume T, Deng K, Hogan BL: Minimal phenotype of mice homozygous for a null mutation in the forkhead/winged helix gene, Mf2. Mol Cell Biol 2000; 20:1419-1425.
198. Cano-Gauci DF, Song HH, Yang H, et al: Glypican-3-deficient mice exhibit developmental overgrowth and some of the abnormalities typical of Simpson-Golabi-Behmel syndrome. J Cell Biol 1999; 146:255-264.
199. Grisaru S, Rosenblum ND: Glypicans and the biology of renal malformations. Pediatr Nephrol 2001; 16:302-306.
200. Grisaru S, Cano-Gauci D, Tee J, et al: Glypican-3 modulates BMP- and FGF-mediated effects during renal branching morphogenesis. Dev Biol 2001; 231:31-46.
201. Hartwig S, Hu MC, Cella C, et al: Glypican-3 modulates inhibitory Bmp2-Smad signaling to control renal development in vivo. Mech Dev 2005; 122:928-938.
202. Boute N, Gribouval O, Roselli S, et al: NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome [In Process Citation]. Nat Genet 2000; 24:349-354.
203. Ciani L, Patel A, Allen ND, French-Constant C: Mice lacking the giant protocadherin mFAT1 exhibit renal slit junction abnormalities and a partially penetrant cyclopia and anophthalmia phenotype. Mol Cell Biol 2003; 23:3575-3582.
204. El-Aouni C, Herbach N, Blattner SM, et al: Podocyte-specific deletion of integrin-linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol 2006; 17:1334-1344.
205. Lavoie JL, Lake-Bruse KD, Sigmund CD: Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule. Am J Physiol Renal Physiol 2004; 286:F965-F971.
206. Sepulveda AR, Huang SL, Lebovitz RM, Lieberman MW: A 346-base pair region of the mouse gamma-glutamyl transpeptidase type II promoter contains sufficient cis-acting elements for kidney-restricted expression in transgenic mice. J Biol Chem 1997; 272:11959-11967.
207. Rubera I, Poujeol C, Bertin G, et al: Specific cre/lox recombination in the mouse proximal tubule. J Am Soc Nephrol 2004; 15:2050-2056.
208. Nelson RD, Stricklett P, Gustafson C, et al: Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. Am J Physiol 1998; 275:C216-C226.
209. Srinivas S, Goldberg MR, Watanabe T, et al: Expression of green fluorescent protein in the ureteric bud of transgenic mice: A new tool for the analysis of ureteric bud morphogenesis. Dev Genet 1999; 24:241-251.
210. Shao X, Somlo S, Igarashi P: Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 2002; 13:1837-1846.
211. Zhu X, Cheng J, Gao J, et al: Isolation of mouse THP gene promoter and demonstration of its kidney-specific activity in transgenic mice. Am J Physiol Renal Physiol 2002; 282:F608-F617.
212. Moeller MJ, Kovari IA, Holzman LB: Evaluation of a new tool for exploring podocyte biology: Mouse Nphs1 5′ flanking region drives LacZ expression in podocytes. J Am Soc Nephrol 2000; 11:2306-2314.
213. Wong MA, Cui S, Quaggin SE: Identification and characterization of a glomerular-specific promoter from the human nephrin gene. Am J Physiol Renal Physiology 2000; 279:F1027-F1032.
214. Moeller MJ, Sanden SK, Soofi A, et al: Two gene fragments that direct podocyte-specific expression in transgenic mice. J Am Soc Nephrol 2002; 13:1561-1567.
215. Engleka KA, Gitler AD, Zhang M, et al: Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev Biol 2005; 280:396-406.
216. Li J, Chen F, Epstein JA: Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis 2000; 26:162-164.
217. Patterson LT, Pembaur M, Potter SS: Hoxa11 and Hoxd11 regulate branching morphogenesis of the ureteric bud in the developing kidney. Development 2001; 128:2153-2161.
218. Mesrobian HG, Sulik KK: Characterization of the upper urinary tract anatomy in the Danforth spontaneous murine mutation. J Urol 1992; 148:752-755.
219. Bullock SL, Fletcher JM, Beddington RS, Wilson VA: Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev 1998; 12:1894-1906.
220. Moser M, Pscherer A, Roth C, et al: Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Dev 1997; 11:1938-1948.
221. Norwood VF, Morham SG, Smithies O: Postnatal development and progression of renal dysplasia in cyclooxygenase-2 null mice. Kidney Int 2000; 58:2291-2300.
222. Kyttala M, Tallila J, Salonen R, et al: MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet 2006; 38:155-157.
223. Weiher H, Noda T, Gray DA, et al: Transgenic mouse model of kidney disease: insertional inactivation of ubiquitously expressed gene leads to nephrotic syndrome. Cell 1990; 62:425-434.
224. Harvey SJ, Jarad G, Gunningham J, et al: Disruption of glomerular basement membrane charge through podocyte-specific mutation of agrin does not alter glomerular permselectivity. Am J Pathol 2007; 171:139-152.
225. Hinkes B, Wiggins RC, Gbadegesin R, et al: Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 2006; 38:1397-1405.
226. Galeano B, Klootwijk R, Manoli I, et al: Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest 2007; 117:1585-1594.
227. Fierlbeck W, Liu A, Coyle R, Ballermann BJ: Endothelial cell apoptosis during glomerular capillary lumen formation in vivo. J Am Soc Nephrol 2003; 14:1349-1354.
228. Sachs N, Kreft M, van den Bergh Weerman MA, et al: Kidney failure in mice lacking the tetraspanin CD151. Cell Biol 2006; 175:33-39.