Paula Krosky
Melinda Hollingshead
Robert Shoemaker
Edward A. Sausville
This chapter provides an overview of the preclinical phase of the development of any anticancer drug, which encompases three overall stages. The first stage is the discovery of a new molecule with therapeutic potential and the selection of the optimal candidate or limited set of candidates for further evaluation. These studies ideally would allow an assessment of how the new molecule differs from currently available therapeutic agents. In the second stage, early preclinical development is directed at increasing confidence that the drug lead will actually function as a useful therapeutic agent in humans. These studies focus predominantly on eliciting activity in animal models of cancer, and ideally correlate the degree of antitumor activity with the pharmacology of the drug. Late preclinical development, the final stage, leads to the completion of regulatory requirements for entry into initial human clinical trials, particularly safety testing at various doses in animal species. Within the past 10 years, the approach to the discovery and development of cancer drugs has undergone a marked change, from focusing mainly on empirical antiproliferative activity as a basis for initial interest in a compound, to selecting drug candidates on the basis of their capacity to modulate molecular targets that are important in cancer pathophysiology.
CANCER DRUG TARGET SELECTION
Our current understanding of cancer cell biology leads to the view that by the time a cancer is clinically manifest in a patient, a number of genetic lesions have occurred in the tumor, resulting in discrete sets of abnormalities that may differ in detail from tumor to tumor, but exist as categories of molecular defects common to essentially all tumor types. This idea, articulated elegantly by Hanahan and Weinberg,1 points to deregulated proliferation-control pathways, loss of tumor suppressor gene function, loss of functions that would promote tumor cell programmed death (apoptosis), acquisition of limitless replication potential through telomere-replicating strategies, activation of host angiogenesis, and the capacity to invade into normal stroma as attributes of all tumors. To these features must be added the capacity of tumors to thwart the host immune system (e.g., Uttenhoeve et al.2). Each of the molecules creating the altered cellular state that underlies tumor cell biology as compared with normal could conceivably be a target for cancer drug discovery and development. Indeed, extensive efforts to define potential targets with altered expression patterns in tumors have been made available through publicly funded (e.g., the Cancer Genome Anatomy Project; see http://www.ncbi.nlm.nih.gov/ncicgap/) and privately maintained databases. These catalogs of genes expressed differentially in tumors are the starting point for current drug discovery and development campaigns. A major question is, how might these targets be prioritized?
One point of view is that those molecules consistently mutated in the course of the development of a cancer in a patient are defined by nature as important in the pathophysiology of that tumor. These molecules may come to attention not only by point mutations in their coding sequence, but by their proximity to frequently observed chromosomal breakpoints or regions of DNA amplification in tumors. Examples of molecules of this type that have been already validated as cancer drug targets include the p210bcr/abl oncoprotein of chronic myelogenous leukemia3 and the HER-2-neu tyrosine kinase4 that is frequently detected by genomic amplification in breast carcinoma. Approved drugs directed at these targets include imatinib and the monoclonal antibody trastuzumab, respectively. In seeming validation of this way of thinking, patients enjoying the best clinical response to the epidermal growth factor receptor tyrosine kinase inhibitor gefitinib have recently been demonstrated to possess with high frequency a mutated, activated form of the agent's target kinase.5, 6 Extending this general approach, the most important targets for cancer drug discovery would be those that can be clearly defined as being mutated in the course of carcinogenesis, or are “downstream” from a mutated molecule and transmit the effects of this mutation to a pathway. These would be exemplary of pathogenic targets because the targets can be reasonably construed as relating to the pathogenesis of the neoplastic process.
Ontologic targets relate to the normal tissue of origin of the tumor. Examples of validated targets of this type include the estrogen or androgen receptors in breast or prostate cancer, respectively, and the CD20 cell surface determinant of non-Hodgkin's lymphoma. Pharmacologic targets relate to the handling or response to a drug itself. For example, dihydropyrimidine dehydrogenase is a target whose degree of activity modulates susceptibility and toxicity of fluorinated pyrimidines, or dexrazoxane modulates levels of free iron and thus alters the cardiotoxic potential of anthracyclines. Stromal or microenvironmental targets include the large array of molecules responsible for sculpting tumor stroma and supporting cell framework including mediators of angiogenesis. The recent regulatory approval of bevacizumab, a monoclonal antibody to vascular endothelial growth factor (VEGF),7 has validated this category of target and promises to be the harbinger of significant discovery efforts around targets of this class. Immunologic strategies, including immunomodulating cytokines or even vaccines, might be considered as special cases ultimately directed at the tumor microenvironment.
The argument for a particular target is strengthened when a phenotype related to tumor behavior can be modulated in cell models (antisense, small interfering RNA, dominant negatives, or ribozyme approaches) or in animal models (knock-outs or transgenics) by genetic approaches to altering the presence or function of a tumor target. For example, topoisomerase IIIβ knock-out mice exhibit premature senescence, which suggests that the use of a topoisomerase poison, such as Camptosar (irinotecan), would encourage rapidly dividing cells toward senescence.8
Other practical criteria that influence the suitability for selection of a molecular target for a drug discovery campaign include the availability (and cost) of reagents to allow screening of leads and the identification of an assay format that is amenable to high-throughput screening (HTS). Finally, the size of the patient population (market) and availability of effective treatments for a particular cancer are usually considered before investing resources toward a new target.
ANTICANCER DRUG SCREENING
Initial recognition of a lead compound can come from a purely molecularly targeted screen, directed against a purified enzyme or a cell engineered to overexpress or underexpress a particular target, or from a cell- or animal-model–based antiproliferative screen, against naturally occurring or engineered tumor cells. Each screening model has its distinct advantages and disadvantages. The molecular targeted approach may yield a drug candidate with clear selectivity for a particular target, but then target modulation must be documented in a cellular context, hopefully with evidence of useful antitumor activity. Ideally, constant evidence of effects “on target” can be an important aid to lead optimization. Cell-based screens have the advantage that drug candidates defined by this route select themselves for the ability to distribute across plasma membranes and survive in the intracellular milieu. On the other hand, their mechanism of action must be determined prior to efficient lead optimization, and cell-based screens are less frequently amenable to high-throughput approaches. In contrast, molecular targeted screens have the attraction of being very amenable to the screening of large collections of molecules.
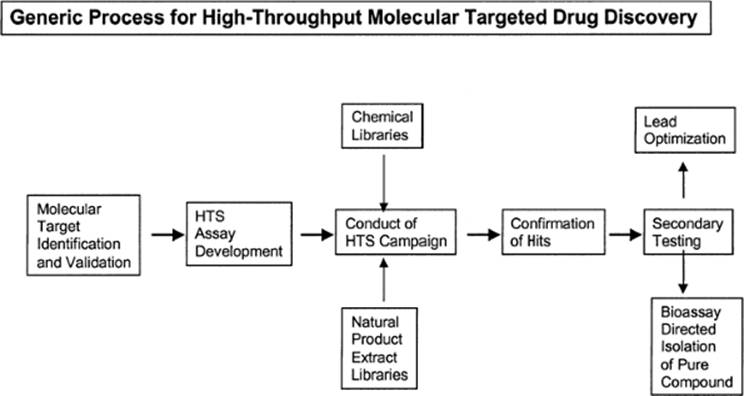
Figure 2.1 illustrates the generic process of high-throughput, molecular targeted screening. The process requires initial identification and validation of a target, followed by development and characterization of an assay suitable for HTS. This assay is then used to screen chemical collections or “libraries” to identify active samples that are the focus of additional testing to establish potency, selectivity, and other features important for further development. Screening is typically conducted in a “campaign” mode, with primary screening data from a particular library evaluated after the whole library has been tested. Criteria for activity are frequently established such that a “hit rate” on the order of 1% is obtained. Thus, for a library of 100,000 samples, 1,000 of the most active samples would be selected for confirmatory testing. Efficient testing usually demands that the primary screen is conducted at a single concentration. Confirmatory testing may be done using the same protocol or may be done in concentration-response fashion to facilitate subsequent consideration of screening leads in the context of potency.
SOURCES OF DIVERSITY FOR LEAD IDENTIFICATION
A crucial issue in any screening project for the purpose of searching for new anticancer drugs is the acquisition of compound libraries. Whether the initial screen is a target-based biochemical screen or an empirical antiproliferation screen, the greater the structural diversity in the set of molecules examined, the more likely a novel inhibitor will be identified. Historically, natural products—defined here as extracts from plants, microbial, and animal sources—have provided an excellent source of bioactive molecules with novel structures and mechanisms of action. In cancer treatment, natural products constitute several major therapeutic classes including the vinca alkaloids, camptothecins, and taxanes. Natural product extracts as sources for cancer drug discovery have been extensively reviewed9 and are not discussed further here. More recently, collections or libraries of synthetic compounds have gained prominence in anticancer drug discovery efforts.
|
|
|
Figure 2.1 Generic process for high-throughput molecular targeted drug discovery. |
Again, there are distinct advantages to each type of compound source. Synthetic compound libraries are either already existing as single compounds or are usually amenable to assignment of activity to a unique chemical structure by a facile deconvolution algorithm. On the other hand, they are limited by the pharmacophore(s) around which the library has been constructed. Although natural product extracts have the possibility of remarkable diversity and stereochemically unique scaffolds, in some cases they require reacquisition from rather exotic ecological niches, optimization of fermentation or extraction approaches, and deconvolution of the active molecule(s) in the extract before the lead can be meaningfully pursued. These strengths and limitations, must be remembered when selecting sources for screening endeavors.
The number and variety of synthetic compounds available for screening endeavors has been magnified by the use of combinatorial chemistry. The method of deliberately synthesizing more than one compound as a result of a single reaction began approximately 20 years ago with the synthesis by Geysen et al.10 of multiple peptides on polyethylene rods. These early endeavors, confined to the synthesis of small peptide libraries, were of limited value to cancer drug discovery because peptides tend not to enter cells and generally are not suitable drug candidates. Subsequent advances in the field included more efficient ways to track the compounds, use of a greater number of scaffolds on which to construct libraries, and use of a wider variety of reagents and amenable reaction conditions. Now, combinatorial chemistry provides an efficient method of exploring chemical space in a focused manner and, when applicable, an excellent means of rapidly defining structure activity relationships around active compounds. Virtually all pharmaceutical companies use combinatorial chemistry at some point in their drug discovery and development programs. The precise position at which combinatorially derived molecules should be employed in a drug discovery and development campaign is a matter of current discussion in the field. “Brute force” screening of millions of compounds not previously enriched for likely valuable molecules has been of little convincing value. On the other hand, combinatorial approaches could greatly enhance the efficiency with which the potential of a lead structure with initial evidence of value is optimized.
Advances in computer-based analysis of the physical properties and interactions of a novel, active compound have provided powerful tools for the pursuit of new anticancer drugs. Medicinal chemists traditionally have used parameters such as steric bulk, hydrogen-bonding ability, and hydrophobic interactions in the design of new drugs. The target pharmacophore now can be further refined by supplementing the information from these physical properties with biologic factors such as the structural biochemistry of the target enzyme or receptor, the nature of the ligand, and the mechanism of the target-ligand interaction when defining the target pharmacophore. Moreover, computer programs such as the Universal Library (Sphinx pharmaceuticals, Durham, NC), Icepick (Axys pharmaceuticals, San Francisco, CA), and Matrix (ComGenex, Inc., Budapest, Hungary) can create “virtual libraries,” which can be evaluated for how thoroughly biochemical, functional space and chemical, diversity space are covered before the proposed structures are synthesized. The use of computer models to design and filter novel structures can be a very efficient mechanism to increase the odds of identifying a potent and selective inhibitor of a well-defined target.
COMPOUND LIBRARIES
Over the years, collections of pure compounds and natural product extracts have coalesced into libraries, some of which are publicly available and arrayed into 96-well plates or 384-well plates in anticipation of HTS. Popular commercial libraries are available from ChemBridge Corp. (San Diego, CA), Maybridge (Cornwall, England), and Sigma-Aldrich (St. Louis, MO; notably the Library of Pharmacologically Active Compounds or LOPAC). The National Cancer Institute (NCI) compound repository is comprised of approximately 600,000 samples obtained by the Developmental Therapeutics Program (DTP) over the past 50 years for use in anticancer drug screening. However, approximately half of the database was submitted under confidentiality agreements that preclude disclosure of information regarding structure or biologic activity. Data for the fraction of the repository for which disclosable information is available, the NCI Open Source Repository (OSR), of approximately 250,000 samples can be obtained at the DTP website (http://www.dtp.nci.nih.gov). Additional smaller collections of compounds are available from an assortment of other suppliers.
A major challenge for all suppliers of compounds is authentication of the structure and quantification of the purity of library materials. Most collections show evidence of the toils of time, storage conditions, and questionable provenance. Most samples in the NCI compound repository were donated by academic chemists or industrial organizations, and were accepted without further chemical characterization. Samples were stored at room temperature unless other conditions were specified by the supplier. As a result, the samples in this collection range in quality from those with a very high degree of sample integrity (purity and authenticity of structure) to those with little or none of the substance indicated by the supplier and reflected in the database records. It is often easier to verify the structure and purity of selected “active” compounds than it is to characterize an entire library.
Numerous algorithms exist by which the diversity of a set of compound structures can be measured; however, little agreement is found regarding the best approach. In general, these software programs partition libraries into a uniform array of blocks or cells on the basis of descriptor coordinates, and the number of cells is proportional to the level of diversity. Some algorithms define atom pair fingerprints, which indicate the presence or absence of pairs of atom types separated by a defined number of bonds, and use them to describe and differentiate each structure. Other algorithms cluster structures into groups; the Jarvis-Patrick clustering algorithm requires that each member of a cluster have in common a predefined number of chemical neighbors.11 The similar Hodes clustering model has been used by the NCI to assess structural novelty of new compounds submitted for screening.12 Often, the goal of these computer algorithms is to define a library of the smallest number of compounds that covers the greatest diversity of space. Using these approaches, large collections of compounds can be winnowed into smaller sublibraries. However, detractors point out that by using these, for the most part, unproven tools and limiting the compounds screened, one risks decreasing the chance of finding drug leads. On the other hand, the economics of HTS calls for prudent use of reagents and encourages the use of such approaches.
A detailed analysis of the NCI OSR was conducted recently, with special emphasis on the uniqueness of the library relative to commercial libraries and other databases.13 Substantial chemical diversity is present in the NCI repository, which has generated of a subset of the OSR containing approximately 140,000 compounds arrayed into multiwell plates. The inventory for most of the remaining compounds is simply too small for these compounds to be provided for routine screening campaigns. Notwithstanding, the plated version of the NCI OSR is a unique publicly available (see http://www.dtp.nci.nih.gov) resource that includes many compounds that cannot be found elsewhere.
In an attempt to provide the structural diversity of the OSR in a smaller package, a further subset of the compounds was selected using Chem-X (Chemical Design, Ltd., Oxfordshire, UK), a program that addresses coverage of three-dimensional space. The goal was to distill the broad set of structures in the larger OSR into a more easily manipulated set of compounds for initial application to screening campaigns. Additional information on the resulting approximately 2,000-member NCI “Diversity Set” may be found at the DTP website. Subsequent analysis of the two-dimensional structural diversity of this subset of compounds was performed by plotting the compounds onto a self-organizing map of “clusters” constructed from information about all of the compounds in the OSR. Although the Diversity Set does have representation spanning the entire map of structural space, it could potentially be more homogenously distributed across the different clusters of structures present in the OSR, and it substantially overrepresents several clusters.14 The NCI currently is selecting a second generation of the Diversity Set that will be optimized for coverage of the chemical space defined by the self-organizing map. Despite these concerns, the current Diversity Set has found application in pilot-scale screening to support HTS assay development and has yielded interesting lead compounds from a variety of molecular targeted screens.15, 16, 17
As with the larger libraries that are commercially available, questions regarding the quality of the compounds with the Diversity Set arise. To address these issues, a chemical analysis of the Diversity Set was performed recently by DTP. A relatively high-throughput high-performance liquid chromatography/mass spectrometry method was devised that accommodates the majority of chemical species. By this analysis, approximately 1,600 of the 1,990 samples in the Diversity Set were shown to contain a mass ion corresponding to the database structure. Purity ranged from >95% to approximately 15% for samples yielding a correct mass ion. Some of the samples for which structural authenticity could not be confirmed have a structure indicated by the database for which this high-throughput analytical approach is not appropriate. Thus, these compounds may have “failed” for technical reasons. Structural authenticity of approximately 300 samples was not supported by this analysis. Detailed information for individual samples is available at the DTP website to allow interpretation of screening results obtained with the Diversity Set.
HIGH-THROUGHPUT SCREENING ASSAY DESIGN
To screen for modulators of a given molecular target, the activity of the protein or the system must be linked to a readily detectable readout. Commonly used types of screening assays can be loosely divided into two categories: separation-based, in which starting material must be removed before the product can be detected, and homogeneous, in which separation of the starting material and the product is not required. Examples of separation-based protocols include filter-binding assays in which the product is selectively retained by the filter (usually an ion-exchange media) and precipitation assays during which one component is selectively precipitated and removed by a glass fiber filter. When used as a screening assay, enzyme-linked immunosorbent assays are generally considered to be separation based. Some techniques that are classified as homogeneous assays include fluorescence polarization, fluorescent resonance energy transfer, scintillation proximity, and luminescent proximity (APLHA Screen). Other homogeneous assays measure changes in light absorbance (UV/vis), in the activity of luciferase, or in the expression of green fluorescent or other related proteins. In general, the throughput of samples with homogeneous assays is much higher than with separation-based assays because the latter require more extensive manipulation of the samples.
Within these categories, the assays can be further divided into cell-based and in vitro (cell-free). Cell-based assays measure the ability of a compound to affect a target within the milieu of an intact cell, whereas cell-free assays measure inhibition of a purified protein. A strength of cell-based assays is the ability to detect inhibitors of an entire targeted pathway (as opposed to a particular step in a pathway). Limitations of cell-based assays include the need to subsequently define the identity of the actual target within the pathway, interference from toxic compounds, and inability to test compounds that cannot penetrate the cell (although this latter feature can also be viewed as an advantage). In comparison, cell-free screening assays are limited neither by cell permeability of compounds nor by nontargeted compound toxicity. When possible, it can be very informative to screen a selected target in both cell-based and in vitro assay systems because their strengths are complementary.
COMMON ISSUES IN HIGH-THROUGHPUT SCREENING IMPLEMENTATION
A major restriction to the development of new HTS assays has been the acquisition and standardization of reagents. Even with substantial miniaturization, many molecularly targeted screens require much more target protein (and/or many more cells) than is needed for basic research on the target. It is essential that sufficient reagents are available for the entire screening effort to avoid batch-to-batch variation. Also, many of the newer, highly sensitive technologies, although affordable on a small scale, become prohibitively expensive when screening large libraries. The availability and cost of reagents can be the pivotal factor in deciding whether to screen a selected target.
An ideal assay plate design includes untreated, negative control wells and wells with compound or condition known to affect the target (positive controls). These provide clear definition of the maximum and minimum signal on an individual plate and, therefore, the window in which compound activity can be measured. However, when studying a newly identified and incompletely characterized target, a specific inhibitor (or activator) may not be known. For some targets, a more generic method of inhibition can be used in the absence of a specific inhibitor (for example, EDTA inhibits most metal-dependent enzymes.) The reproducibility among the control wells within a plate and the reproducibility of the control wells among the plates within a set are indicative of the quality of the screen; this will be addressed later.
Once a new screening assay has been designed and standardized, it is important to characterize its performance by completing pilot-scale screens. For DTP screens, the NCI Training Set of 230 compounds routinely is tested at least twice, and the correlation between the two sets of data provides a quantitative measure of the reproducibility of the assay.14 Sometimes intriguing lead structures can emerge from pilot-scale screening efforts; for example, a recent screen of the NCI Diversity Set for inhibitors of the HIF1α pathway identified several camptothecin analogs, the activities of which have been confirmed in secondary testing.17
EXAMPLES FROM NCI DTP SCREENS
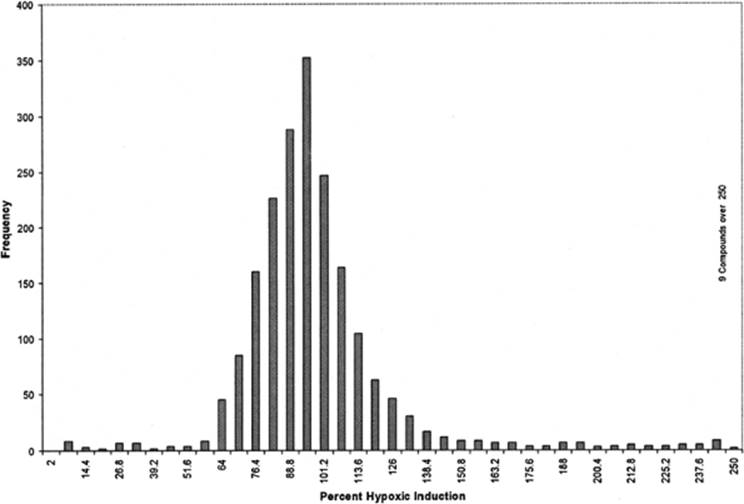
The DTP website contains searchable and downloadable primary HTS data (Diversity Set) from screens targeting Met signaling, hypoxic cell signaling, the CEBPα transcription factor, and HIV-1 nucleocapsid/nucleic acid interaction. The first three of these utilized cell-based assays, whereas the latter was a cell-free assay. Figures 2.2 and 2.3 illustrate the character of the HTS data from the standpoint of frequency of active samples and the potential to utilize one screen as a selectivity “counterscreen” for another. Screening data from these two campaigns can be combined and compared to assess selectivity of the active compounds. The plot shown in Figure 2.4 demonstrates facile identification of samples that were active in both screens and thus of reduced interest as screening leads. For example, the single data point in the lower right corner of Figure 2.4, which had striking induction in the CEBPα-luciferase reporter assay and notable inhibition in the HIF-1α-luciferase reporter assay, was a quinocarmycin analog that appears to activate CEBP as a stress response and to inhibit HIF by generation of DNA damage.
CHEMOINFORMATICS
Lead Identification and Development
The goal of any molecularly targeted screen is to identify inhibitors (or activators, in some cases) of a particular protein or pathway, and it is a sine qua non of such exercises to distinguish real “hits” from false-positives. Two primary factors to consider are the overall quality of the screening data and the definition of a hit. The most common metric to quantify the quality of screening data is known as Z' and is defined by the following equation18:

In this equation, µ is the mean value of the positive and negative controls for the assay and σ is the standard deviation of these values. Assays for which Z' > 0.5 are considered acceptable for HTS. There are three commonly used strategies for selecting active compounds for secondary analysis: (a) to select all compounds displaying a particular level, or range of activity; (b) to select all compounds with activity that falls outside a designated level of deviation from the mean; and (c) to select a particular number of compounds defined by the institutional limitations for follow-up. In practice, the third strategy is employed, to some degree, with all screens because there are always practical limits on follow-up capabilities. Consistency among control samples and a manageable number of hits are evidence of a well-designed screen and increases the probability of identifying high-quality lead compounds.
|
|
|
Figure 2.2 Distribution of activity observed in primary HTS of the NCI Diversity Set (1 µM) for inhibition of induction of hypoxic cell signaling. The majority of samples tested were without inhibitory activity, with a modal activity around 100%. A minority of inhibitory samples are observed in the left-hand tail of distribution. This tail also includes toxic and nonselective inhibitors of transcription that were resolved in secondary testing. Data and details of the screening protocol are available on the DTP website. Overall results are described in Rapisarda et al.17 |
|
|
|
Figure 2.3 Distribution of activity observed in primary HTS of the NCI Diversity Set (1 µM) for induction of CEBPα-luciferase reporter. Only a very small number of samples in the right side of the distribution demonstrated activity equal to 50% or more of the positive control (retinoic acid). Data and details of the screening protocol are available on the DTP website. |
The process for culling hits is multifold and usually begins by confirming compound identity and activity and demonstrating a dose-response relationship. Ideally, the proposed mechanism of action of compounds identified in in vitro assays is corroborated in cell-based assays and vice versa. Often, confirmed active compounds subsequently are filtered through one or more algorithms designed to recognize druglike molecules and purge compounds known to be toxic, reactive, and/or to affect multiple molecular targets nonselectively. Filtering software packages are available from several companies, including Leadscope, Inc. (Columbus, OH), Accerlys, Inc. (Burlington, MA), Bioreason, Inc.
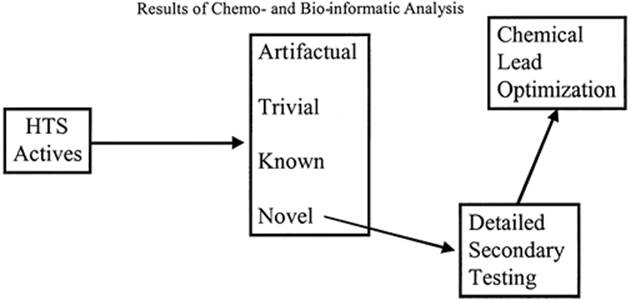
(Santa Fe, NM), and Golden Helix, Inc. (Bozeman, MT). Eventually, the active, desirable compounds are used as the basis for lead optimization and development. At a practical level, this process can be viewed as an informatic triage of the screening data in which active compounds or classes are partitioned into categories, as illustrated in Figure 2.5.
|
|
|
Figure 2.4 Plot of inhibitory activity in the HIF-1α HTS of the NCI Diversity Set versus activation of the CEBPα-luciferase reporter. Compounds in the lower right quadrant meet the indicated criteria for activity (reduction of HIF-1α signaling to 50% or less of the control value and induction of the CEBPα-luciferase reporter to at least 50% of the level of the positive control compound). |
|
|
|
Figure 2.5 Results of chemoinformatic and bioinformatic analysis. |
Pharmacokinetic Considerations
At some point during the culling process, the pharmacologic demands of absorption, distribution, metabolism, and excretion must be considered. A common criticism and limitation of HTS is that such screening assays are usually designed to find the compound that is most potent and most selective against a particular target, without regard to solubility, bioavailability, and/or toxicity. Historically, considerable effort in drug discovery programs has been spent refining drug leads that subsequently fail in the development process because of toxicity, lack of in vivo efficacy, or poor pharmacokinetic properties.
The development of predictive, inexpensive, in vitro assays and computer models to quantify pharmacologic properties are helping to alleviate this lead development bottleneck and substantially enhancing the multidisciplinary character of drug discovery. For example, assays such as those developed by Exelexis, Inc. (San Francisco, CA) use parameters including partition coefficients, P-glycoprotein efflux, P-450 induction and metabolism, and protein binding to predict the pharmacokinetics of novel compounds. These assays have been standardized to the greatest extent possible with the use of in vivo data from animals and humans. There are numerous, commercially available software packages that rely entirely on “well-trained” computer algorithms to predict compound absorption (GastroPlus [Simulations Plus, Inc., Lancaster, CA] and iDEA pk Express [LION Bioscience Inc., Cambridge, MA]), subcompartment penetration, plasma protein binding, metabolism (MetabolExpert [CompuDrug International Inc., Sedona, AZ], META [Multicase, Beachwood, OH], and Meteor LHASA, Dept. of Chem, Leeds, UK), and drug-drug interactions (Q-DIPS Dr Pascal Bonnabry, Geneva, Switzerland). The use of these and other similar model systems is permitting analysis of the pharmacologic properties of a larger number of compounds earlier in the development process; it is hoped that this will reduce the rates of compound failure.
Ideally, initial pharmacologic studies in animals would provide confidence that the area under the concentration × time curve (AUC) achievable in animals approximates the drug exposure that is necessary in tumor cells propagated in vitro to achieve an effect on the target molecule leading to an expected tumor-modulating effect. This is most frequently assessed as proliferation, but could also be related to AUCs causing a decrease in clonogenic potential in semisolid media, an assay that has been related to tumorigenic or metastatic capacity.
NATIONAL CANCER INSTITUTE CELL-BASED SCREENING SYSTEMS
Anticancer drug screening began at the NCI about 50 years ago in response to Congressional interest in removing barriers to anticancer drug discovery and development. NCI screening initially was an empirical process in which test samples were administered to mice bearing transplantable tumors. The L1210 and P388 mouse leukemia models proved to be highly sensitive and evolved into prescreening models supplemented with secondary testing in solid mouse tumors and xenografts. Detailed historical information on the in vivo screening program can be found in Zubrod.19 Additional details, as well as the rationale for transition to in vitro human tumor cell models, can be found in Shoemaker and Sausville.20
This latter model, comprising 60 tumor cell lines representing multiple histologic tumor types, was operated as a primary antitumor drug screen for over a decade and was particularly noteworthy for the information-rich character of the screening data. A recent review has described in detail the value of this screen for characterizing mechanisms of growth inhibition and cytotoxicity.21 Consideration of the 60-cell screening data for a new chemotype, even one selected on the basis of an a priori defined molecular target, can reveal important aspects of the action of the new molecule. This includes its utilization of previously described detoxification mechanisms such as the P-glycoprotein efflux pump, the relation of compound action to conventional cytotoxics, or tying the compound action to the presence or function of molecular targets or pathways in tumor cells. These interpretations of the 60-cell screening data utilize the COMPARE, neural network, cluster analysis, and self-organizing map algorithms.21, 22, 23 However, the potential of the 60 tumor cell line screen has been limited by the labor-intensive nature of cell production, plating, drug addition, and end point processing. This, in addition to the growing number of attractive molecular targets for drug discovery, has led to development of an infrastructure for high-throughput molecular targeted screening at the NCI, much of which has been previously described.
IN VIVO EFFICACY TESTING
At the completion of the primary screening process, including confirmation of hits, definition of lead structures and their structural optimization (ideally using information derived from binding of the lead to its intended target molecule), a limited set of well-understood compounds should have been characterized for preliminary pharmacology and chemical stability in the physiologic milieu. Sufficient quantity should be available to allow detailed understanding of their actions in animal models of tumor cell growth, or in vivo efficacy evaluations. Historically, this testing has been empirically utilized with compounds tested at maximum tolerated doses on fixed schedules established for individual tumor models.24 In the era of molecular targeted drug development, customization of in vivo model testing for individual agents is becoming more common, using tumor cells selected to overexpress or underexpress the drug's target. Indeed, the availability of a pharmacodynamic assay that can support testing to establish an optimal dose for target modulation can be crucial to development of a new agent. Data relating antitumor activity to pharmacodynamic and pharmacokinetic effects potentially can be followed from the preclinical arena to the earliest clinical testing. Lack of target modulation in the clinic or failure to obtain or maintain adequate drug levels would be an early indication of lack of therapeutic potential. Conversely, clear evidence of target modulation at drug levels associated with preclinical activity can argue strongly for accelerated clinical development. Recent examples of approved drug molecules that have utilized modulation of their intended molecular target through the development process as key evidence supporting and informing their continued development include the proteosome inhibitor bortezomib25 and the epidermal growth factor antagonist gefitinib.26
A common concern is how valid are various animal models of cancer drug action in projecting ultimate clinical activity. Recently, Johnson et al.27 have examined the utility of subcutaneous xenografts in athymic mice in this regard for cytotoxic agents introduced between 1980 and 1990. It is clear that such models are relatively poor in predicting activity on a disease-by-disease basis. For example, activity in breast cancer xenografts poorly predicts ultimate activity in human breast cancer. On the other hand, activity in many different models does predict a higher likelihood of activity in some human clinical population. Specifically, activity of the agent in >33% of the models tested predicted an approximately 50% likelihood of clinical activity in at least one human tumor type. In contrast, cytotoxic agents with <33% of the tumor models affected had essentially no evidence of clinical activity. Careful consideration of the achievable pharmacology in humans in designing experiments in mice can clearly increase the predictive value of the murine experiments.28 Whether these statistics can be improved by selection of models created to depend on the function of the molecular target of the action of a drug remains to be seen, and is the object of much current research interest.
PRACTICAL ASPECTS OF IN VIVO EFFICACY TESTING
A variety of animal models for evaluating anticancer therapies have been described. Although each model has its protagonists, no single model is ideal for all applications. Thus, one must determine what information is desired from the in vivo study to make an informed choice regarding which model(s) to use. Table 2.1 lists the types of critical information needed from efficacy studies.
Initial Detection of Antitumor Activity: The Hollow Fiber Assay
The desire to implement a high throughput, low-cost, in vivo prescreen so that lead compounds could be prioritized for efficacy testing in the xenograft models led to development of the hollow fiber assay.29 This assay has emerged as the initial in vivo screen by the NCI for lead compounds with cytostatic or cytocidal potential. Compounds with activity in the hollow fiber assay are considered for testing in human tumor xenografts after compound mechanism of action, formulation, and pharmacokinetic issues are considered. The hollow fiber assay allows screening of ≥50 compounds per week in a 10-day assay that uses <500 mg of compound while testing for growth suppression of less than 106 tumor cells.29,30 The assay as practiced at NCI evaluates the activity of a test agent against a standard panel of 12 cell lines consisting of 2 lines each from the breast, colon, lung, melanoma, CNS, and ovarian tumor histology subpanels. For the standard assay, compounds are evaluated at two dose levels based on the maximum tolerated dose (MTD) determined in mouse toxicity assays. The high test dose is set at (1.5 × MTD)/4 and the low dose is 67% of the high test dose. Hollow fiber cultures of each cell line are prepared in vitro and implanted subcutaneously and intraperitoneally into mice. Because compound delivery is accomplished through intraperitoneal injection, the anticellular activity can be assessed in a same site (intraperitoneally/intraperitoneally) and a distant site (subcutaneously/intraperitoneally) modality in the same mouse. The mice are treated with vehicle or test agent for 4 days, and the fibers are retrieved for evaluation of viable cell mass using a formazan dye conversion assay. The percent net growth of each cell line is calculated by comparison with a set of control fibers assessed for viable cell mass on the day of implantation.
|
TABLE 2.1 INFORMATION TO BE GAINED FROM IN VIVO EFFICACY TESTING |
|
|
Rodent Tumor Models: Additional Considerations
A variety of tumor models have been described in rodents, with those of greatest interest occurring in rats and mice. This results partly from their relatively low body weights, which minimize the amount of chemotherapeutic needed, and the fact that their physiology is the best understood of the rodent species and there are many inbred strains available. Murine leukemias, grown in the peritoneal cavity and producing morbidity as an end point, have been used for many years. Use of the L1210 and P388 leukemias in drug screening efforts was reviewed by Waud.31 In addition to the leukemias, a variety of solid tumors are available for modeling antitumor activity. These include tumors of varying histologic types and implant sites. These tumor systems have been thoroughly reviewed by Corbett et al.32
Human Tumor Xenograft Models
There is much in the literature regarding human tumor xenograft models that rely on immunocompromised mice as hosts for tumors of various histologies.24,33, 34, 35, 36 These models can be divided into subgroups based on the site at which the tumor cells are inoculated, as well as the end point measured by the assay. Tumors have been successfully generated following tumor cell inoculation into the peritoneal cavity, the subcutaneous tissues, the vascular network via cardiac or tail vein puncture, under the renal capsule, and various other organ sites.
One of the simplest xenograft models involves direct injection of tumor cells into the peritoneal cavity with subsequent administration of test agent by one of several routes. In most cases, the end point for these intraperitoneal xenograft assays is host morbidity. This results from ascites and intraperitoneal tumor formation and dissemination of the tumor to distant organs, for example, the brain, for some tumor cell lines. Because there is no requirement for daily tumor measurements, these assays are less labor-intensive and less cost-prohibitive than other xenograft models. Unfortunately, not all cells are amenable to growth in the peritoneal cavity, so the desired cell line must be assessed for its growth potential. An intraperitoneal tumor is difficult to observe until ascites are present, so it is important to standardize the assay so that all of the inoculated mice do produce viable tumor growth. It is also of value to determine the minimum inoculum required to give the desired end point in all of the test mice (e.g., time to ascites development). The NCI in vivo screening program has found the HL-60 promyelocytic leukemia, U251 glioblastoma, LOX melanoma, and several other ovarian, leukemic, and lymphoma cell lines acceptable for subcutaneous tumor models (M Hollingshead, unpublished data). With these models, direct administration of the test agent into the peritoneal cavity offers the greatest potential for activity for most compounds because they do not have to achieve effective plasma concentrations or distribute to the target tissues. This approach is often referred to as a “same-site” model because the target (tumor cells) and the test agent are placed into close proximity. Although this does not address issues such as agent uptake, distribution, metabolism, and excretion, it does allow an initial assessment of the in vivo potential of the test article. The occurrence of protein binding, local cellular uptake, rapid systemic absorption and distribution, metabolism, and host toxicity can be preliminarily assessed in these same-site models. Nonetheless, activity only in a same-site model is less valuable than activity that occurs after a molecule traverses several phamacologic compartments.
|
TABLE 2.2 SUBCUTANEOUS XENOGRAFT EFFICACY PARAMETERS |
|
|
Subcutaneous Xenograft Tumors
The subcutaneous xenograft model is commonly used for chemotherapeutic assessment as the tumor growth can be monitored visually throughout the experiment. For these assays, tumor cells are placed under the skin of immunocompromised mice (e.g., SCID, nu/nu, NIH-III, SCID/bg, SCID/NOD) and the cells are monitored for growth by daily observation. When tumors are detected at the inoculation site, daily measurement of the tumor volume can be accomplished with calipers. Generally, the tumor length and width are measured, and the tumor volume is calculated using one of several formulas.37 The NCI assumes the tumor is a prolate ellipsoid so the volume = [tumor length (mm) × tumor width (mm)2]/2. Assuming a unit volume of 1, then the volume in cubic millimeters is equal to the tumor weight in milligrams. The tumor weights versus time can be plotted to produce tumor growth curves that are compared between control and experimentally treated mice to ascertain whether treatment has an impact on tumor growth. Parameters for measuring compound efficacy in these subcutaneous xenograft models have been described elsewhere. The parameters used in the NCI drug-screening program are given in Table 2.2, and a summary of the implementation features of various models are summarized in Table 2.3.
Orthotopic Implants
One of the deficiencies of the subcutaneous xenograft model is its failure to produce the metastatic lesions that ultimately kill many cancer patients.38, 39There are a few subcutaneous xenograft models that do produce metastatic lesions (e.g., LOX melanoma, SK-MEL-28 melanoma, MDA-MB-435 breast, DU-145 prostate), but they are the exception rather than the rule.
Fidler and colleagues40 and Fidler41 demonstrated that the occurrence of metastatic lesions is not the result of a random event. Rather, they are selective, and the metastatic event consists of a series of steps that depend on the tumor cell injection site. Fidler demonstrated that a tumor implanted orthotopically, for example, into the organ of origin, behaved more like the clinical disease and was thus a better model for human cancer than the subcutaneous models. This concept has proven useful for studying tumor biology and therapies because orthotopic implantation generates tumors that produce metastatic lesions similar to those seen in man.42, 43, 44 To further extend the importance of orthotopic xenografts, Fidler and coworkers45 demonstrated differences in drug sensitivity between orthotopically and subcutaneously implanted tumor tissue. Although tissues of any origin can be implanted orthotopically, those most commonly used are breast, colon, brain, melanoma, and lung tumors. Building on the concept of orthotopic implantation, Hoffman46 developed and patented the MetaMouse. This model differs from other orthotopic models in that the implanted tumor material consists of surgically attached pieces of tumor rather than cell suspensions or simple trocar-implanted tumor fragments. The tumor fragments used in the MetaMouse are obtained directly from human patients or from human tumor xenografts grown subcutaneously on immunocompromised mice. Giavazzi47 and Hoffman39 have reviewed orthotopic models and the resulting metastatic lesions.
Metastatic Models
Metastasis, the process by which tumor cells leave their tissue of origin and colonize distant tissues, has become a target for new antineoplastic therapies. The metastatic process consists of multiple events that result from invasion of the tissue and of vascular and lymphatic components adjacent to the primary tumor. This process is initiated by basement membrane invasion resulting from proteolysis and cell motility. Following tumor cell intravasation into the circulation, tumor cell emboli are trapped in distant capillary beds in which extravasation can occur.48 A small percentage of these embolic tumor cells may survive to produce tumors.49 Another critical component in these events is the tumor vasculature, as it provides the tumor with nutrients for growth as well as provides a pathway by which tumor cells can gain entry to distant tissues. Each step in the metastatic process may be viewed as a potential target to interrupt tumor spread.50 Unfortunately, the in vivo tumor models currently available do not assess the individual targets but rely on demonstration of an effect downstream of these targets, an antitumor effect. This can be measured with standard subcutaneous xenograft models if the target is applicable, such as antiangiogenic agents. If the target is not relevant in the subcutaneous models, such as basement membrane proteolysis, then the orthotopic models are an option. These models are generally metastatic, so an effect on one or more steps in the metastatic process should reduce the number of detectable tumor nodules at the metastatic site(s).
|
TABLE 2.3 TYPES OF MURINE IN VIVO EFFICACY MODELS |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
Intravenous Tumor (Disseminated Tumor)
Disseminated tumor models in which intravenously injected tumor cells colonize the lungs and other tissues offer another method for evaluating agents effective in the later stages of metastasis. Perhaps the best known of the disseminated tumor models is the murine melanoma, B16-BL6.51 The B16-BL6 melanoma is able to metastasize from a primary subcutaneous site as well as following intravenous injection. Thus, the same tumor cell can be used to assess both upstream and downstream events in the metastatic process. This model has been utilized successfully to evaluate a matrix metalloproteinase inhibitor designed to inhibit basement membrane degradation.52 Although not as well characterized as B16-BL6, there are human xenograft models in which intravenously administered cells produce disseminated disease in immune-deficient mice, particularly SCID mice. Examples of human tumor cell lines producing this effect are LOX melanoma, SK-MEL-28 melanoma, K562 chronic myelogenous leukemia, AS-283 AIDS-related lymphoma, and A549 lung tumor.53Problems with the disseminated models include the need for reproducible intravenous inoculations in rodents and the inability to identify the exact cause for reductions in lung or other organ colonization.
Antiangiogenic Agents
The impact of inhibiting angiogenesis on tumor growth and metastasis has led to the development of specific antiangiogenic assays in addition to the standard tumor growth-inhibition assays. Various in vitro assays can assess the impact of a therapeutic agent on endothelial cell proliferation, migration, and cord formation.54 These assays help delineate the mechanism of action for a potential therapeutic, but they may not show activity with all agents. Compounds whose effects are mediated through a secondary mechanism, such as cytokine induction, would not demonstrate effects in these in vitro assays. For in vivo studies, many laboratories use the chicken chorioallantoic membrane as a substrate to assess antiangiogenic agents.55 This is a more complex assay than the in vitro assays, but it does lack several features of human neoplasia. These differences include (a) it is not mammalian, (b) it is embryonic, (c) it does not simulate the tumor angiogenesis microenvironment, (d) it is only semiquantitative, and (e) some researchers believe that it may not measure clinically relevant activity.
Various in vivo models are described that measure the growth of blood vessels into an exogenously administered substrate. Although various substrates have been described,54 the most commonly used is Matrigel to which various angiogenic agents, e.g. VEGF, bFGF, have been added.56 Matrigel (BD Biosciences, San Jose, CA) is a basement membrane extract in which new blood vessels develop following injection into the subcutaneous tissue of rodents. By quantitating the number of vessels and/or the hemoglobin content, the angiogenesis response can be defined. Of note, Matrigel can be used to support xenogeneic tumor cells for injection into mice because it protects the tumor cells, provides a physiologic support, and may provide a medium into which vascular components can migrate.
The corneal angiogenesis assay provides another tumor-independent assay.57, 58 For this assay, controlled-release pellets containing angiogenic agents, for examply, basic fibroblast growth factor (bFGF) or VEGF, are placed into corneal micropockets and vessel growth is quantified in the presence or absence of treatment with putative antiangiogenic agents. This approach was used in the initial assessment of the antiangiogenic potential of TNP-470 and thalidomide, two purported antiangiogenic compounds taken into clinical trials
Molecular Targets and Transgenic Animals
One current interest in cancer therapeutics involves modulation of various molecular targets in neoplastic cells. These targets include oncogenes, which promote unregulated cell growth (e.g., ras, fos, myc, sis, erb) and tumor suppressor genes that suppress tumor growth (e.g., p53).
Although a large number of targets have been defined, the importance of each of them is an area of current research. The interest in these targets has led to a two-pronged strategy to develop animal models to validate these molecular targets, both as important tumorigenicity targets and as chemotherapeutic targets. One approach is to transform cells with oncogenes so that the effect of the oncogene on cellular activity can be assessed both in vitro and in vivo. If the nontransformed cell line is tumorigenic, then the in vivo activity of a compound against the transformed and nontransformed cells can be compared using methods described earlier. Another approach is the generation of transgenic mice bearing one or more mutations. In many instances these transgenic mice develop spontaneous tumors at a defined age.
The impact of chemotherapeutic agents on tumor development and growth may be assessed following treatment at various times relative to the predicted tumor occurrence. The range of models available has recently been reviewed.59 As an example of their value, Barrington et al.60 reported that L-744,832, a farnesyltransferase inhibitor, is p53-independent utilizing transgenic mice expressing one or more oncogenes in the presence or absence of p53. These transgenic mice offer an exciting approach to manipulating potential treatment targets; however, their use for routine in vivo screening is often limited by the time required for tumor development and the amount of compound necessary to treat for a protracted period of time. Additionally, the number of mice developing tumors may be <50%, so extremely large treatment groups are necessary to obtain statistically relevant results. For example, a transgenic model in which only 30% of mice develop tumors may require hundreds of test animals to achieve statistical validity. It is recommended that a statistician be consulted to aid in determining the appropriate treatment group size for a given tumor model prior to embarking on a chemotherapy trial.
NEW IMAGING TECHNOLOGIES IN EFFICACY STUDIES
Highly sensitive imaging techniques for quantitating fluorescent and bioluminescent signals in live rodents are providing new approaches to model development efficacy. Tumor cell lines or transgenic mice engineered to express green fluorescent protein, luciferase, or other fluorescent/bioluminescent markers can be imaged in vivo. This allows small numbers of tumor cells to be visualized and quantitated following orthotopic implantation. Furthermore, small metastatic lesions, not obvious in classic tumor models, can be visualized in essentially all tissue sites. Transgenic mice can be engineered to express these signals when tumors are initiated or when other promoters are activated. This provides a means to progressively monitor otherwise “hidden” events. Even though these technologies are in the early phases of validation, they offer possibilities for significant enhancement of rodent tumor models.61 Although these techniques are amenable to rodent models, they do not currently offer a potential for translating into direct human clinical endpoints. However, commercial availability of rodent positron emission tomography, magnetic resonance imaging, and computed tomography scanners is opening a pathway to techniques that can be directly translated into human studies. This is important not only for improving diagnostic imaging models, but for assessing the impact of treatment on tumor blood flow, tumor mass, and other clinically relevant end points.
PRECLINICAL PHARMACOLOGY AND TOXICOLOGY STUDIES
Following clear demonstration of antitumor activity in an appropriate range of animal models, the next phase of a drug's preclinical development addresses its effects on the host organism, independent of the presence of tumor. The goal of preclinical toxicology studies for anticancer drugs is to determine in appropriate species the maximally tolerated dose (MTD), the nature of dose-limiting toxicity (DLT), demonstration of schedule-dependent toxicity, and the reversibility of that toxicity.62, 63, 64, 65, 66 This results in the ability to estimate a “safe” starting dose for initial clinical trials, which for small organic molecules is one-tenth the MTD, or one-third the toxic dose low (TDL) in nonrodents. The TDL is defined as the highest toxic dose that, when doubled corresponds to a nonlethal dose. Thus, one-sixth is used with the highest nonlethal dose. In no case is a dose that just begins to elicit reversible toxicity utilized to define the starting dose.
Toxicologic evaluations are typically divided into preliminary or “range-finding” studies, which assess clinical characteristics of drugs administered at a range of doses selected to bracket those that are effective and those that are toxic. More detailed investigational new drug (IND)-directed toxicology evaluations focus on the actual proposed clinical use schedule and seek to define a nontoxic, just toxic, and overtly toxic series of doses during which animals are followed for the appearance and reversibility of clinical signs, along with studies of clinical chemistry, hematology, and histopathology of all organs.63 These studies are currently required by the Food and Drug Administration (FDA) to utilize two species, including at least one nonrodent species, and to utilize the proposed clinical dose and schedule.64, 65, 66 In contrast, evaluation of “biologicals” including antibodies, immunotoxins, and vaccines, generally utilize only the most relevant animal species, exposed to the agent using the anticipated clinical route and schedule.
Previously, toxicology evaluations utilized “standard” protocols; for example, the standard NCI toxicology protocols from 1980 to 1988 included determination of the LD10 on day 1 and day 1 through day 5 dosing schedules, followed by assessment of safety and DLT when administered at the LD10 on a day 1 and day 1 through day 5 schedule.65 The current development paradigm, in contrast, is “agent-directed” and targets the most effective route and schedule of administration.63, 64, 65, 66 For example, twice weekly, or continuous infusion for periods as long as 28 days are now routine initial phase 1 schedules.
Preclinical analytical studies routinely define a suitable assay for study of bulk and formulated drug stability and a separate assay for the drug in biologic fluids. Pharmacologic studies are conducted to determine the pharmacokinetics in rats and dogs after single intravenous doses and by the route and schedule that mimics what was efficacious in animal models. Toxicokinetic studies, referring to the relationship between plasma drug levels and the elicitation of toxicity in animals, are conducted as part of the toxicology studies. Additional studies that increase confidence that the drug will perform well in clinical trials provide demonstration that one can obtain efficacious drug levels in vivo, with correlation of drug plasma levels, and/or AUC with safety and toxicity. It should be noted that the toxicologic studies previously described are required for all drugs, whereas the development of pharmacology assays are only pursued if possible. Not all antineoplastic agents have been amenable to assay at the time of entry into clinical trial (especially many highly potent natural products), so pharmacology information is desirable, but not legally essential for clinical study.
INITIATION OF CLINICAL STUDIES
The regulatory requirements for use of an agent vary with the type of use. To enter early clinical trials (e.g., phase 1 or 2), documentation that the proposed schedule may be safely given is the goal of FDA review. To allow marketing of the agent, FDA must determine that the agent is safe and effective. The latter is customarily defined by behavior in phase 3 trials, in which the test agent is compared with standard or no therapy. An increasingly popular strategy is to attempt to gain “accelerated approval” for an agent to treat a dire or life-threatening disease. In that event, phase 2 data may be sufficient to allow such accelerated approval if the following end points were obtained: unequivocal evidence of tumor diminution or clear documentation of preserved performance status in the absence of a “response” including favorable outcome of “quality of life” indicators. In the event of accelerated approval, postmarketing stipulations to assure continued judgment of likely benefit are defined.
To allow inception of phase 1 dose-escalation trials, following completion of toxicologic evaluations, reversible toxicity must be demonstrated to be likely after the first manifestation of a drug's adverse effects (as opposed to poorly predictable and irreversible toxicity). The drug is then ready for entry into the clinic, provided that a reliable and workable formulation of the agent has been defined. To allow commencement of the clinical trial, an IND application must be approved by the FDA. The components of an IND include cover sheet (Form 1571); table of contents; introductory statement; general investigative plan; investigator's brochure; initial clinical protocol; chemistry, manufacturing, and control (this extremely important section provides for the precise description and chemical characteristics of the drug under study, how it was made, and how it is to be labeled); toxicology data; pharmacology data (if available); previous human experience; and miscellaneous (including potential for abuse, and results of radiotracer experiments). Responsibilities of the sponsor of phase 1 studies include submission of the IND application, assuring qualifications of investigators, writing and securing local Institutional Review Board (IRB) approval of protocols, shipping investigational agents and maintaining detailed shipping records corresponding to lots sent, assessing adverse drug reactions and submitting them in a timely fashion to FDA, preparing an annual report of the IND activities to the FDA, monitoring quality of the data through periodic audits, assuring that the use and disposition of the investigational agent is properly accounted for at the study sites, assuring informed consent is obtained for each patient entering into the study, tracking amendments made to protocols after inception of clinical studies, and informing investigators of new information pertinent to the trial. These regulations are contained in 21 CFR 312.50.
REFERENCES
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57–70.
2. Uttenhoeve C, Pilotte L, Théate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nature Med 2003;9: 1269–1274.
3. O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic myelogenous leukemia. N Engl J Med 2003;348:994–1004.
4. Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 2002;20: 719–726.
5. Paez JG, Janne PA, Lee PC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1458–1461.
6. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350: 2129–2139.
7. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorecal cancer. N Engl J Med 2004;350:2335–2342.
8. Kwan KY, Wang JC. Mice lacking DNA topoisomerase IIIbeta develop to maturity but show a reduced mean lifespan. Proc Natl Acad Sci U S A 2001;98:5717–5721.
9. Cragg GM, Newman DJ. Antineoplastic agents from natural sources: achievements and future directions. Expert Opin Investig Drugs 2000;9:2783–2797.
10. Geysen HM, Meloen RH, Barteling SJ. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc Natl Acad Sci U S A, 1984;81:3998–4002.
11. Jarvis RA, Patrick EA. Clustering using a similarity measure based on shared near neighbors. IEEE Trans Comput 1973;C22: 1025–1034.
12. Hodes L. Clustering a large number of compounds. 1. Establishing the method on an initial sample. J Chem Inf Comput Sci 1989;29:66–71.
13. Voigt JH, Bienfait B, Wang S, et al. Comparison of the NCI open database with seven large chemical structural databases. J Chem Inf Comput Sci 2001;41:702–712.
14. Shoemaker RH, Scudiero DA, Melillo G, et al. Application of high-throughput, molecular-targeted screening to anticancer drug discovery. Curr Top Med Chem 2002;2:229–246.
15. Stephen AG, Worthy KM, Towler E, et al. Identification of HIV-1 nucleocapsid protein: nucleic acid antagonists with cellular anti-HIV activity. Biochem Biophys Res Commun 2002;296:1228–1237.
16. Lazo JS, Aslan DC, Southwick EC, et al. Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J Med Chem 2001;44: 4042–4049.
17. Rapisarda A, Uranchimeg B, Scudiero DA, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res 2002;62:4316–4324.
18. Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67–73.
19. Zubrod CG. Origins and development of chemotherapy research at the National Cancer Institute. Cancer Treat Rep 1984;68:9–19.
20. Shoemaker RH, Sausville EA. New drug development. In: Souhami RL, Tannock I, Honenberger P, et al., eds. Oxford Textbook of Oncology. Oxford: Oxford University Press, 1999: 781–788.
21. Holbeck SL. Update on NCI in vitro drug screen utilities. Eur J Cancer 2004;40:785–793.
22. Paull KD, Shoemaker RH, Hodes L, et al. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst 1989;81:1088–1092.
23. Rabow AA, Shoemaker RH, Sausville EA, et al. Mining the National Cancer Institute's tumor-screening database: identification of compounds with similar cellular activities. J Med Chem 2002;45:818–840.
24. Plowman J, Dykes DJ, Hollingshead MG, et al. Human tumor xenograft models in NCI drug development. In: Teicher BA, ed. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval., Totowa, NJ: Humana Press, 1997:101–125.
25. AdamsJ, Kauffman M: Development of the proteosome inhibitor Velcade. (Bortezomib). Cancer Invest 2004;2:304–11.
26. El-Rayes BF, LoRusso PM: Targeting the epidermal growth factor receptor. Br J Cancer 2004;91:418–24.
27. Johnson JI, Decker S, Zaharevitz D, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer 2001;84:1424–1431.
28. Peterson JK, Houghton PJ. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur J Cancer 2004;40:837–844.
29. Hollingshead MG, Alley MC, Camalier RF, et al. In vivo cultivation of tumor cells in hollow fibers. Life Sci 1995;57:131–141.
30. Decker S, Hollingshead M, Bonomi CA, et al. The hollow fibre model in cnacer drug screening: the NCI experience. Eur J Cancer 2004;40:821–826.
31. Waud WR. Murine L1210 and P388 Leukemias. In: Teicher BA, ed. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Totowa, NJ: Humana Press, 1997: 59–74.
32. Corbett T, Valeriote F, LoRusso P, et al. In vivo methods for screening and preclinical testing use of rodent solid tumors for drug discovery. In: Teicher BA, ed. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Totowa, NJ: Humana Press, 1997:75–99.
33. Fiebig HH, Maier A, Burger AM. Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anti-cancer drug discovery. Eur J Cancer 2004;40:802–820.
34. Giovanella BC, Stehlin JS. Heterotransplantation of human malignant tumors in “nude” thymusless mice. I. Breeding and maintenance of “nude” mice. J Natl Cancer Inst 1973;51:615–619.
35. Leonessa F, Green D, Licht T, et al. MDA435/LCC6 and MDA435/LCC6MDR1: ascites models of human breast cancer. Br J Cancer 1996;73:154–161.
36. McLemore TL, Abbott BJ, Mayo JG, et al. Development and application of new orthotopic in vivo models for use in the US National Cancer Institute's drug screening program. In: Wu B-q, Zheng J, eds. Immune-Deficient Animals in Experimental Medicine. 6th International Workshop of Immune-Deficient Animals. Beijing: Basel, 1989:334–343.
37. Clarke R. Issues in experimental design and endpoint analysis in the study of experimental cytotoxic agents in vivo in breast cancer and other models. Breast Cancer Res Treat 1997;46:255–278.
38. Fine DL, Shoemaker R, Gazdar A, et al. Metastasis models for human tumors in athymic mice: useful models for drug development. Cancer Detect Prev Suppl 1987;1:291–299.
39. Hoffman RM. Patient-like models of human cancer in mice. Curr Perspec Molec Cell Oncol 1992;1:311–326.
40. Fidler IJ, Wilmanns C, Staroselsky A, et al. Modulation of tumor cell response to chemotherapy by the organ environment. Cancer Metastasis Rev 1994;13:209–222.
41. Fidler IJ. Rationale and methods for the use of nude mice to study the biology and therapy of human cancer metastasis. Cancer Metastasis Rev 1986;5:29–49.
42. Giavazzi R, Jessup JM, Campbell DE, et al. Experimental nude mouse model of human colorectal cancer liver metastases. J Natl Cancer Inst 1986;77:1303–1308.
43. Mohammad RM, Al-Katib A, Pettit GR, et al. An orthotopic model of human pancreatic cancer in severe combined immunodeficient mice: potential application for preclinical studies. Clin Cancer Res 1998;4:887–894.
44. Berry KK, Siegal GP, Boyd JA, et al. Development of a metastatic model for human endometrial carcinoma using orthotopic implantation in nude mice. Intl J Oncol 1994;4:1163–1171.
45. Wilmanns C, Fan D, O'Brian CA, et al. Orthotopic and ectopic organ environments differentially influence the sensitivity of murike colon carcinoma cells to doxorubicin and 5-fluroaracil Int J Cancer 1992;52:98–104.
46. Hoffman RM. Fertile seed and rich soil. In: Teicher BA, ed. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Totowa, NJ: Humana Press, 1997: 127–144.
47. Giavazzi R. Metastic models. In: Boven E, Winograd B, eds. The Nude Mouse in Oncology Research. Boca Raton, FL: CRC Press, 1991:117–132.
48. Dickson RB, Johnson MD, Maemura M, et al. Anti-invasion drugs. Breast Cancer Res Treat 1996;38:121–132.
49. Fidler IJ. Selection of successive tumour lines for metastasis. Nat New Biol 1973;242:148–149.
50. Zetter BR. Angiogenesis and tumor metastasis. Annu Rev Med 1998;49:407–424.
51. Talmadge JE, Fidler IJ. Cancer metastasis is selective or random depending on the parent tumour population. Nature 1982;297: 593–594.
52. Chirivi RG, Garofalo A, Crimmin MJ, et al. Inhibition of the metastatic spread and growth of B16-BL6 murine melanoma by a synthetic matrix metalloproteinase inhibitor. Int J Cancer 1994; 58:460–464.
53. Guilbaud N, Kraus-Berthier L, Saint-Dizier D, et al. Antitumor activity of S 16020-2 in two orthotopic models of lung cancer. Anticancer Drugs 1997;8:276–282.
54. Taraboletti G, Giavazzi R. Modelling approaches for angiogenesis. Eur J Cancer 2004;40:881–889.
55. Schlatter P, Konig MF, Karlsson LM, et al Quantitative study of intussusceptive capillary growth in the chorioallantoic membrane (CAM) of the chicken embryo. Microvasc Res 1997;54:65–73.
56. Passaniti A, Taylor RM, Pili R, et al. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab Invest 1992;67:519–528.
57. Muthukkaruppan V, Auerbach R. Angiogenesis in the mouse cornea. Science 1979;205:1416–1418.
58. Kenyon BM, Voest EE, Chen CC, et al. A model of angiogenesis in the mouse cornea. Invest Ophthalmol Vis Sci 1996;37:1625–1632.
59. Hansen K, Khanna C. Spontaneous and genetically engineered animal models: use in preclinical cancer drug development. Eur J Cancer 2004;40:858–880.
60. Barrington RE, Subler MA, Rands E, et al. A farnesyltransferase inhibitor induces tumor regression in transgenic mice harboring multiple oncogenic mutations by mediating alterations in both cell cycle control and apoptosis. Mol Cell Biol 1998;18:85–92.
61. Hollingshead MG, Bonomi CA, Borgel SD, et al. A potential role for imaging technology in anticancer efficacy evaluations. Eur J Cancer 2004;40:890–898.
62. Grieshaber CK. Agent-directed preclinical toxicology for new antineoplastic drugs. In: Valeriote FA, Corbett H, eds. Cytotoxic Anticancer Drugs: Models and Concepts for Drug Discovery and Development. Boston: Kluwer Academic Publishers, 1992: 247–260.
63. Tomaszewski JE, Smith AC. Safety testing of antitumor agents. In: Williams PD, Hottendorf GH, eds. Comprehensive Toxicology, Toxicity Testing and Evaulation., Oxford: Elservier Science Ltd., 1997:299–309.
64. DeGeorge JJ, Ahn CH, Andrews PA, et al. Regulatory considerations for preclinical development of anticancer drugs. Cancer Chemother Pharmacol 1998;41:173–185.
65. Lowe MC, Davis RD. The current toxicology portocol of the National Cancer Institute. In: Hellman D, Carter S, eds. Fundamentals of Cancer Chemotherapy. New York: McGraw Hill, 1987:228–235.
66. Tomaszewski JE. Multi-species toxicology approaches for oncology drugs: the US perspective. Eur J Cancer 2004;40:907–913.