ORGANIZATIONAL PRINCIPLES OF THE CNS
The brain is a complex assembly of interacting neurons and nuclei that regulate their own and each other’s activities in a dynamic fashion, generally through chemical neurotransmission. It is useful to examine the major anatomical regions of the CNS and their associations with specific neurotransmitter systems and the effect of pharmacological agents thereon.
CELLULAR ORGANIZATION OF THE BRAIN
Neurons. Neurons are classified according to function (sensory, motor, or interneuron), location, the identity of the transmitter they synthesize and release or the class or classes of receptor expressed on the cell surface. Neurons exhibit the cytological characteristics of highly active secretory cells with large nuclei: large amounts of smooth and rough endoplasmic reticulum; and frequent clusters of specialized smooth endoplasmic reticulum (Golgi complex), in which secretory products of the cell are packaged into membrane-bound organelles for transport from the perikaryon to the axon or dendrites. The sites of interneuronal communication in the CNS are termed synapses. Like peripheral “junctions,” central synapses are denoted by accumulations of tiny (50-150 nm) synaptic vesicles. The proteins of these vesicles have specific roles in transmitter storage, vesicle docking, and secretion and re-storage of transmitter.
Support Cells. Neurons in the CNS are outnumbered by support cells, including macroglia, microglia, vascular elements, the cerebrospinal fluid (CSF)-forming cells of the choroid plexus found within the intracerebral ventricular system, and the meninges, which cover the surface of the brain and comprise the CSF-containing envelope. Macroglia are the most abundant support cells; some are categorized asastrocytes (cells interposed between the vasculature and the neurons, often surrounding individual compartments of synaptic complexes). Astrocytes play a variety of metabolic support roles including furnishing energy intermediates and supplementary removal of neurotransmitters following release. The oligodendroglia, a second prominent category of macroglia, are myelin-producing cells. Myelin, made up of multiple layers of compacted membranes, insulate segments of axons bioelectrically and permit nondecremental propagation of action potentials. Microglia are derived from mesoderm and are related to the macrophage/monocyte lineage. Some microglia reside within the brain, while additional cells of this class may be recruited to the brain during periods of inflammation following either microbial infection or brain injury.
BLOOD-BRAIN BARRIER. The blood-brain barrier (BBB) is an important boundary between the periphery and the CNS that forms a permeability barrier to the passive diffusion of substances from the bloodstream into the CNS. The BBB diminishes the rate of access of many chemicals from plasma to the brain and is the localization of several drug export systems in the cells that constitute the BBB (see Chapter 5). An exception exists for lipophilic molecules, which diffuse fairly freely across the BBB and accumulate in the brain.
This barrier, nonexistent in the peripheral nervous system, is much less prominent in the hypothalamus and in several small, specialized organs (the circumventricular organs) lining the third and fourth ventricles of the brain: the median eminence, area postrema, pineal gland, subfornical organ, and subcommissural organ.
Selective barriers to permeation into and out of the brain also exist for small, charged molecules such as neurotransmitters, their precursors and metabolites, and some drugs. These diffusional barriers are viewed as a combination of the partition of solute across the vasculature (which governs passage by definable properties such as molecular weight, charge, and lipophilicity) and the presence or absence of energy-dependent transport systems (see Chapter 5). Important exceptions are the specific uptake transporters for amino acids, one of which contributes to the therapeutic utility of L-dopa in the treatment of Parkinson disease.
The brain clears metabolites of transmitters into the fluid-containing lateral ventricles by excretion via the acid transport system of the choroid plexus. Substances that rarely gain access to the brain from the bloodstream often can reach the brain when injected directly into the cerebrospinal fluid. Under certain conditions, it may be possible to open the BBB, at least transiently, to permit the entry of chemotherapeutic agents. Cerebral ischemia and inflammation also modify the BBB, increasing access to substances that ordinarily would not affect the brain.
CHEMICAL COMMUNICATION IN THE CNS
A central concept of neuropsychopharmacology is that drugs that influence behavior and improve the functional status of patients with neurological or psychiatric diseases act by enhancing or blunting the effectiveness of specific transmitters and channels.
Identified targets for centrally acting drugs include ion channels that mediate change in excitability induced by neurotransmitters, neurotransmitter receptors, and transport proteins that reaccumulate released transmitter. The transport proteins include those selective for NE, dopamine, and serotonin (NET, DAT, and SERT) that accumulate released transmitter and those that package it for reuse (e.g., VMAT2). Inhibition of reuptake increases the concentration and dwell time of transmitter in the synaptic space (e.g., as do serotonin selective reuptake inhibitors and cocaine). Inhibition of vesicular storage leads to depletion of releasable neurotransmitter (e.g., inhibition of VMAT2 and NE storage by reserpine).
An understanding at the systems level is required to assemble the structural and functional properties of specific central transmitter systems, linking the neurons that make and release a given neurotransmitter to its behavioral effects. The entire concept of animal models of human psychiatric diseases rests on the assumption that scientists can appropriately infer from observations of behavior and physiology (heart rate, respiration, locomotion, etc.) that the states experienced by animals are equivalent to the emotional states experienced by human beings expressing similar physiological changes.
IDENTIFICATION OF CENTRAL TRANSMITTERS. The criteria for identification of central transmitters require the same data set used to establish the transmitters of the autonomic nervous system (seeChapter 8):
• The transmitter must be shown to be present in the presynaptic terminals of the synapse and in the neurons from which those presynaptic terminals arise.
• The transmitter must be released from the presynaptic nerve concomitantly with presynaptic nerve activity.
• When applied experimentally to target cells, the effects of the putative transmitter must be identical to the effects of stimulating the presynaptic pathway.
• Specific pharmacological agonists and antagonists should mimic and antagonize, respectively, the measured functions of the putative transmitter with appropriate affinities and order of potency.
Many brain and spinal cord terminals contain more than 1 transmitter substance. Coexisting substances (presumed to be released together) may either act jointly on the postsynaptic membrane, or may act presynaptically to affect release of transmitter from the presynaptic terminal. Clearly, if more than 1 substance transmits information, no single agonist or antagonist would be expected to faithfully mimic or fully antagonize activation of a given presynaptic element. Co-storage and co-release of ATP and NE are an example. In addition to being released as a cotransmitter with other biogenic amines, ATP and adenosine have been shown to mediate diverse effects through interactions with purinergic receptors.
MANY NEUROTRANSMITTERS AND DRUGS ACT ON IDENTIFIED NEURONAL MACROMOLECULES
A number of molecular mechanisms link receptor occupancy to biological responses (see Chapter 3). The most commonly seen post-receptor events are changes in ion flux through channels formed by a multi-subunit receptor complex and the alteration of intracellular signaling via transmembrane receptor systems.
ION CHANNELS. The electrical excitability of neurons is achieved through modification of ion channels in neuronal plasma membranes. Na+, K+, and Ca2+, as well as Cl– anions, are regulated in their flow through highly discriminative ion channels. These channels are grouped structurally and termed voltage-dependent channels (Figure 14–1), and Cl–channels (Figure 14–2). Ligand-gated ion channels, regulated by the binding of neurotransmitters, form another distinct group of ion channels; a prominent example is the nicotinic acetylcholine receptor, a Na+ channel when activated (see Figures 11–1 and 11–2).

Figure 14–1 Structural similarities of voltage-dependent Na+, Ca2+, and K+ channels. A. The subunit in both Ca2+ and Na+ channels contains 4 subunits, each with 6 transmembrane hydrophobic domains. The hydrophobic regions that connect segments 5 and 6 in each domain form the pore of the channel. Segment 4 in each domain includes the voltage-sensor. (Adapted with permission from Catterall W.Neuron, 2000;26:13–25. © Elsevier.) B. The Ca2+ channel also requires several auxiliary small proteins (α2, β, γ, and δ); α2 and δ subunits are linked by a disulfide bond. Regulatory subunits also exist for Na+ channels. C. Voltage-sensitive K+ channels (Kv) and the rapidly activating K+ channel (KA) share a similar putative hexaspanning structure resembling in overall configuration 1 repeat unit within the Na+ and Ca2+ channel structure; the inwardly rectifying K+ channel protein (Kir) retains the general configuration of just loops 5 and 6. Regulatory t subunits (cytosolic) can alter Kv channel functions. Voltage-dependent channels provide for rapid changes in ion permeability along axons and within dendrites and for excitation-secretion coupling that releases neurotransmitters from presynaptic sites. The transmembrane Na+ gradient (~140 mM outside vs. ~14 mM inside the cell) means that increases in permeability to Na+ causes depolarization. In contrast, the K+ gradient (~4 mM outside the cell vs. ~120 mM inside) is such that increased permeability to K+ results in hyperpolarization. Changes in the concentration of intracellular Ca2+ (extracellular free Ca2+: 1.25 mM; intracellular Ca2+: resting ~100 nM, rising to ~1 μM when Ca2+ entry is stimulated) affects multiple processes in the cell and are critical in the release of neurotransmitters.

Figure 14–2 Three families of Cl– channel. Due to the Cl– gradient across the plasma membrane (~116 mM outside vs. 20 mM inside the cell), activation of Cl– channels causes an inhibitory postsynaptic potential (IPSP) that dampens neuronal excitability; inactivation of these channels can lead to hyperexcitability. There are 3 distinct types of Cl– channel:
• Ligand-gated channels are linked to inhibitory transmitters including GABA and glycine.
• ClC channels, of which 9 subtypes have been cloned, affect Cl-flux, membrane potential, and the pH of intracellular vesicles.
• Cystic fibrosis transmembrane conductance regulated (CFTR) channels bind ATP and are regulated by phosphorylation of serine residues.
(M, transmembrane domains; NBF, nucleotide-binding fold; R, regulatory [phosphorylation] domain.) (Reproduced with permission from Jentsch J. Chloride channels: A molecular perspective. Curr Opin Neurobiol, 1996;6:303–310. Copyright 3 Elsevier.)
Two other families of channels regulate ion fluxes: cyclic nucleotide–gated (CNG) channels, and transient receptor potential (TRP) channels. CNG channels consist of 2 groups:
• The CNG channels, which play important roles in sensory transduction for olfactory and photoreceptors
• The hyperpolarization-activated, cyclic nucleotide–gated (HCN) channels
HCN channels are cation channels that open with hyperpolarization and close with depolarization; upon direct binding of cyclic AMP or cyclic GMP, the activation curves for the channels are shifted to more hyperpolarized potentials. These channels play essential roles in cardiac pacemaker cells and presumably in rhythmically discharging neurons.
TRP channels are a family of hexaspanning receptors containing a cation-permeable pore. TRP channels respond to multiple stimuli and function in sensory physiology, including thermosensation, osmosensation, and taste. Members of the IRPV subfamily (vanilloid receptors) interact with ligands such as the endogenous cannabinoid anandamide and capsaicin, a “hot” or “irritant” component of chili peppers.
TRANSMEMBRANE RECEPTOR SYSTEMS. A variety of membrane receptors interact with neurotransmitters and neurohormones. These systems have been described in detail inChapter 3.
CELL SIGNALING AND SYNAPTIC TRANSMISSION. Most cell-to-cell communication in the CNS involves chemical transmission that requires a series specializations for transmitter synthesis, storage, release, recognition, and termination of action (Figure 14–3; see also Figures 8–2, 8–3, and 8–5). In addition to primary neurotransmitters (usually vesicular), there are neurohormones neuromodulators, and neurotrophic factors that influence CNS function:
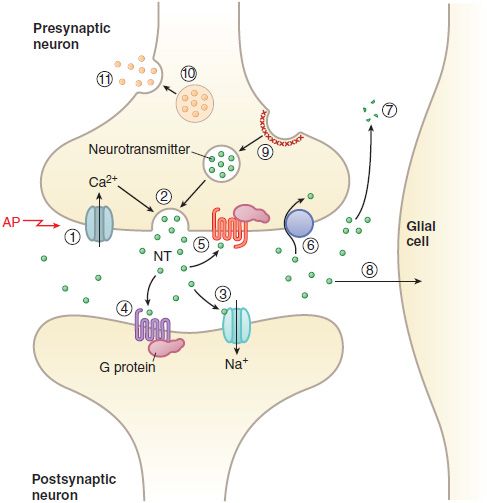
Figure 14–3 Transmitter release, action, and inactivation. Depolarization opens voltage-dependent Ca2+ channels in the presynaptic nerve terminal. (1) The influx of Ca2+ during an action potential (AP) triggers (2) the exocytosis of small synaptic vesicles that store neurotransmitter (NT) involved in fast neurotransmission. Released neurotransmitter interacts with receptors in the postsynaptic membranes that either couple directly with ion channels (3) or act through second messengers, such as (4) GPCRs. Neurotransmitter receptors in the presynaptic nerve terminal membrane (5) can inhibit or enhance subsequent exocytosis. Released neurotransmitter is inactivated by reuptake into the nerve terminal by (6) a transport protein coupled to the Na+ gradient (e.g., for DA, NE, and GABA), or by (7) degradation (ACh, peptides), or by (8) uptake and metabolism by glial cells (Glu). The synaptic vesicle membrane is recycled by (9) clathrin-mediated endocytosis. Neuropeptides and proteins are stored in (10) larger, dense core granules within the nerve terminal. These dense core granules are released from (11) sites distinct from active zones after repetitive stimulation.
NEUROHORMONES. The anterior and posterior pituitary secrete a variety of hormones and releasing factors. Hypothalamic neurons affecting the anterior pituitary release their hormones into the hypothalamic–adenohypophyseal portal blood system, which delivers them to the anterior pituitary, where they regulate the release of trophic hormones (i.e., ACTH, FSH, GH, LH, prolactin) into the blood. Other hypothalamic neurons project onto the posterior pituitary, where they release their peptide contents, oxytocin and arginine vasopressin (antidiuretic hormone) into the systemic circulation (seeChapters 25 and 38 and Figure 38–1).
NEUROMODULATORS. The distinctive feature of a modulator is that it originates from nonsynaptic sites, yet influences the excitability of nerve cells. Substances such as CO and ammonia, arising from active neurons or glia, are potential modulators acting through non-synaptic actions. Similarly, circulating steroid hormones, steroids produced in the nervous system (i.e., neurosteroids), locally released adenosine, and other purines, eicosanoids, and nitric oxide (NO) are regarded as modulators and/or neurotransmitters.
NEUROTROPHIC FACTORS. Neurotrophic factors are substances produced within the CNS by neurons, astrocytes, microglia, or inflammatory or immune cells that assist neurons in their attempts to repair damage. Seven categories of neurotrophic peptides are recognized:
• Classic neurotrophins (nerve growth factor, brain-derived neurotrophic factor, and the related neurotrophins)
• Neuropoietic factors, (e.g., cholinergic differentiation factor [also called leukemia inhibitory factor], ciliary neurotrophic factor, and some interleukins)
• Growth factor peptides, such as epidermal growth factor, transforming growth factors GGand a, glial cell–derived neurotrophic factor, and activin A
• Fibroblast growth factors
• Insulin-like growth factors
• Platelet-derived growth factors
• Axon guidance molecules, some of which also affect cells of the immune system
CENTRAL NEUROTRANSMITTERS
Neurotransmitters can be discussed by chemical categories: amino acids, amines, and neuropeptides. Other substances that may participate in central synaptic transmission include purines (such as adenosine and ATP), NO, and arachidonic acid derivatives.
AMINO ACIDS. The CNS contains uniquely high concentrations of certain amino acids, notably glutamate and β-aminobutyric acid (GABA) that potently alter neuronal firing. They are ubiquitously distributed within the brain and they produce prompt, powerful, and readily reversible but redundant effects on neurons. The dicarboxylic amino acids (e.g., glutamate and aspartate) produce excitation, while the monocarboxylic amino acids (e.g., GABA, glycine, -alanine, and taurine) cause inhibition. Following the emergence of selective antagonists, identification of selective receptors and receptor subtypes became possible. Figure 14–4 shows these amino acid transmitters and their drug congeners.

Figure 14–4 Amino acid transmitters and congeners.
GABA. GABA receptors have been divided into 3 main types:
• The GABAA receptor (the most prominent GABA receptor subtype), a ligand-gated Cl-ion channel, or ionotropic receptor
• The GABAB receptor, a GPCR, or metabotropic receptor
• The GABAC receptor, a transmitter-gated Cl-channel
GABAA receptor has been extensively characterized as the site of action of many neuroactive drugs, notably benzodiazepines, barbiturates, ethanol, anesthetic steroids, and volatile anesthetics (Figure 14–5). The GABAA receptor is probably pentameric or tetrameric in structure, with subunits that assemble around a central pore. The major form of the GABAA receptor contains at least 3 different subunits α, β, and γ, with likely stoichiometry of 2α, 2β, 1α. All 3 subunits are required to interact with benzodiazepines with the profile expected of a native GABAA receptor.

Figure 14–5 Pharmacologic binding sites on the GABAA receptor. (Reproduced with permission from Nestler EJ, Hyman SE, Malenka RC, eds. Molecular Neuropharmacology. New York: McGraw-Hill, 2009, p 135. © 2009 by The McGraw-Hill Companies, Inc.)
The GABAB or metabotropic GABA receptors interact with Gi to inhibit adenylyl cyclase, activate K+ channels, and reduce Ca2+ conductance and with Gq to enhance PLC activity. Presynaptic GABABreceptors function as autoreceptors, inhibiting GABA release, and may play the same role on neurons releasing other transmitters. The GABAC receptor is less widely distributed than the A and B subtypes. GABA is more potent by an order of magnitude at GABAC than at GABAA receptors, and a number of GABAB agonists (e.g., baclofen) and modulators (e.g., benzodiazepines and barbiturates) do not interact with GABAC receptors. GABAC receptors are found in the retina, spinal cord, superior colliculus, and pituitary.
GABA mediates the inhibitory actions of local interneurons in the brain and may also mediate presynaptic inhibition within the spinal cord. GABA-containing neurons frequently coexpress 1 or more neuropeptides.
The most useful compounds for confirmation of GABA-mediated effects have been bicuculline and picrotoxin; however, many convulsants whose actions previously were unexplained (including penicillinand pentylenetetrazol) are relatively selective antagonists of the action of GABA. Useful therapeutic effects have not yet been obtained through the use of agents that mimic GABA (such as muscimol), inhibit its active reuptake (such as 2,4-diaminobutyrate, nipecotic acid, and guvacine), or alter its turnover (such as aminooxyacetic acid).
GLYCINE. Many of the features described for the GABAA receptor family also apply to the inhibitory glycine receptor, which is prominent in the brainstem and spinal cord. Multiple subunits assemble into a variety of glycine receptor subtypes, the complete functional significance of which is not known.
GLUTAMATE AND ASPARTATE. Glutamate and aspartate have powerful excitatory effects on neurons in virtually every region of the CNS. Glutamate receptors are classed functionally either as ligand-gated ion channel (ionotropic) receptors or as metabotropic GPCRs (Table 14–1). The ligand-gated ion channels are further classified into N-methyl-D-aspartate (NMDA) receptors and non-NMDA receptors. The non-NMDA receptors include the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and kainic acid (KA) receptors.
Table 14–1
Classification of Glutamate and Aspartate Receptorsa

NMDA receptor agonists include open-channel blockers such as phencyclidine (PCP or “angel dust”); antagonists include 5,7-dichlorokynurenic acid, which acts at an allosteric glycine-binding site, and ifenprodil, which may act as a closed-channel blocker. The activity of NMDA receptors is sensitive to pH and to modulation by a variety of endogenous agents including Zn2+, some neurosteroids, arachidonic acid, redox reagents, and polyamines such as spermine. NMDA receptors are involved in normal synaptic transmission; activation of NMDA receptors is associated more closely with the induction of various forms of synaptic plasticity rather than with fast point-to-point signaling in the brain.
AMPA and kainate receptors mediate fast depolarization at glutamatergic synapses in the brain and spinal cord. AMPA or kainate receptors and NMDA receptors may be colocalized at many glutamatergic synapses. A well-characterized phenomenon involving NMDA receptors is the induction of long-term potentiation (LTP) and its converse, long-term depression (LTD) (Figure 14–6).
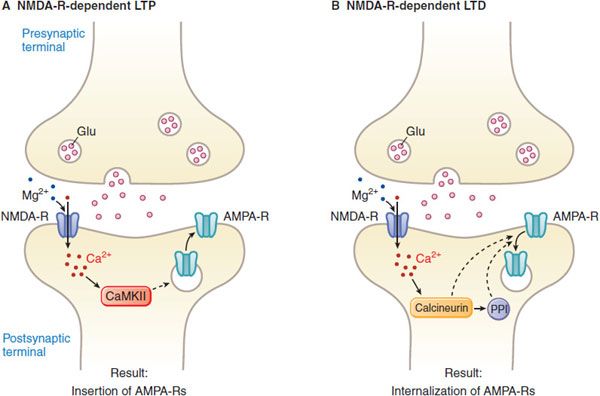
Figure 14–6 NMDA receptor-dependent LTP and LTD. LTP refers to a prolonged (hours to days) increase in the size of a postsynaptic response to a presynaptic stimulus of given strength. Activation of NMDA receptors is obligatory for the induction of LTP that occurs in the hippocampus. NMDA receptors normally are blocked by Mg2+ at resting membrane potentials. Thus, activation of NMDA receptors requires glutamate binding and the simultaneous depolarization of the postsynaptic membrane. This is achieved by activation of AMPA/kainate receptors at nearby synapses involving inputs from different neurons. AMPA receptors also are dynamically regulated to affect their sensitivity to the synergism with NMDA. Thus, NMDA receptors may function as coincidence detectors, being activated only when there is simultaneous firing of 2 or more neurons. LTD is the converse of LTP. A. NMDA receptor-dependent LTP requires post synaptic NMDA receptor activation leading to a rise in Ca2+ and activation of CaM kinase II (CaMKII). AMPA receptor insertion into the postsynaptic membrane is a major mechanism underlying LTP expression. B. NMDA receptor-dependent LTD is triggered by Ca2+entry through postsynaptic NMDA receptor channels, leading to increases in the activity of the protein phosphatases calcineurin and PP1. LTD occurs when postsynaptic AMPA receptors are internalized. (Redrawn with permission from Nestler EJ, Hyman SE, Malenka RC, eds. Molecular Neuropharmacology. New York: McGraw-Hill, 2009, p 132. © 2009 by The McGraw-Hill Companies, Inc.)
GLUTAMATE EXCITOTOXICITY. High concentrations of glutamate lead to neuronal cell death (Figure 14–7). The cascade of events leading to neuronal death is thought to be triggered by excessive activation of NMDA or AMPA/kainate receptors, allowing significant influx of Ca2+ into neurons. Glutamate-mediated excitotoxicity may underlie the damage that occurs after ischemia or hypoglycemia in the brain, during which a massive release and impaired reuptake of glutamate in the synapse leads to excess stimulation of glutamate receptors and subsequent cell death. NMDA receptor antagonists can attenuate neuronal cell death induced by activation of these receptors.

Figure 14–7 Mechanisms contributing to neuronal injury during ischemia-reperfusion-induced glutamate release. Several pathways contribute to excitotoxic neuronal injury in ischemia, with excess cytosolic Ca2+ playing a precipitating role. DAG, diacylglycerol; GluR, AMPA/kainate type of glutamate receptors; IP3, inositol trisphosphate; mGluR, metabotropic glutamate receptor; NMDA- R, N—methyl-D-aspartate receptor; O2–, superoxide radical; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PL, phospholipids, PLA2/C phospholipases, VSCC, voltage-sensitive Ca2+channel. COX, cyclooxygenase; LOX, lipoxygenase; NCX, Na+/Ca2+ exchanger; mtPTP, mitochondrial permeability transition pore. (Reproduced with permission from Dugan LL, Kim-Han JS: Hypoxic-ischemic brain injury and oxidative stress, in Siegel GS, Albers RW, Brady S, Price D, eds. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 7th ed. Burlington, MA: Elsevier Academic Press, 2006, p 564. e 2006, American Society for Neurochemistry.)
ACETYLCHOLINE. The effects of ACh result from interaction with a mixture of nicotinic and muscarinic receptors. Nicotinic ACh receptors (see Figures 11–1 and 11–2) are found in autonomic ganglia, the adrenal gland, and in the CNS. Their activation by ACh results in a rapid increase in the influx of Na+, depolarization, and the activation of voltage-sensitive Ca2+ influx. There are 5 subtypes of muscarinic receptors, all of which are expressed in the brain. M1, M3, and M5 couple to Gq while the M2 and M4 receptors couple to Gi (Table 14–2).
Table 14–2
Subtypes of Muscarinic Receptors in the CNS

CATECHOLAMINES. The brain contains separate neuronal systems that use 3 different catecholamines—dopamine (DA), NE, and epinephrine (EPI). Each system is anatomically distinct and serves separate, functional roles within its field of innervation.
DOPAMINE. More than half the CNS content of catecholamine is DA. The 5 receptors for DA are GPCRs that regulate adenylyl cyclase activity via coupling to Gs (D1 and D5) and Gi (D2-4) (Table 14–3). DA-containing pathways and receptors have been implicated in the pathophysiology of schizophrenia and Parkinson disease and in the side effects seen following pharmacotherapy of these disorders (see Chapters 16 and 22).
Table 14–3
Dopamine Receptors in the CNS

Large amounts of DA are found in the basal ganglia. There are 3 major DA-containing pathways in the CNS: the nigrostriatal, the mesocortical/mesolimbic, and the tuberoinfundibular, as described inChapter 13 and depicted in Figure 13–7.
NOREPINEPHRINE. Both B and adrenergic receptors subtypes are present in the CNS; all are GPCRs (Table 14–4; see also Chapter 8).
Table 14–4
Adrenergic Receptors in the CNS

β receptors couple to Gs and thence to adenylyl cyclase. α1 Adrenergic receptors are coupled to Gq, resulting in stimulation of the PLC-IP3/DAG-Ca2+-PKC pathway, and are associated predominantly with neurons. d1 Receptors on noradrenergic target neurons respond to NE with depolarizing responses because of decreases in K+ conductance. α2 Adrenergic receptors are found on glial and vascular elements, as well as on neurons. They are prominent on noradrenergic neurons, where they presumably couple to Gi, inhibit adenylyl cyclase, and mediate a hyperpolarizing response due to enhancement of an inwardly rectifying K+ channel (via heterodimer). e2 Receptors are located presynaptically where they function as inhibitory autoreceptors. The antihypertensive effects of clonidine may result from stimulation of such autoreceptors.
There are relatively large amounts of NE within the hypothalamus and in certain parts of the limbic system, such as the central nucleus of the amygdala and the dentate gyrus of the hippocampus. NE also is present in significant amounts in most brain regions. Mapping studies indicate that noradrenergic neurons of the locus ceruleus innervate specific target cells in a large number of cortical, subcortical, and spinomedullary fields.
EPINEPHRINE. EPI-containing neurons are found in the medullary reticular formation. Their physiological properties have not been identified.
5-HYDROXYTRYPTAMINE (SEROTONIN; 5HT). There are 5 types of 5HT receptors, with 14 distinct subtypes (Table 14–5). These subtypes exhibit characteristic ligand-binding profiles, couple to different intracellular signaling systems, exhibit subtype-specific distributions within the CNS, and mediate distinct behavioral effects of 5HT. All 5HT receptors are GPCRs coupling to a variety of G-protein t subunits except the 5HT3 receptor, which is a ligand-gated ion channel. The localizations and properties of 5HT receptors are summarized in Table 13–1, their physiological roles in various CNS activities in Table 13–2, the electrophysiological events associated with receptor activation in Table 13–4, and aspects of their clinical pharmacology in Table 13–5.
Table 14–5
5HT Receptors in the CNS

In mammalian CNS, neurons containing 5HT are found in 9 nuclei lying in or adjacent to the midline (raphe) regions of the pons and upper brainstem. Cells receiving cytochemically demonstrable 5HT input, such as the suprachiasmatic nucleus, ventrolateral geniculate body, amygdala, and hippocampus, exhibit a uniform and dense investment of serotonergic terminals.
HISTAMINE. Four subtypes of histamine receptors have been described; all are GPCRs coupling to regulate adenylyl cyclase and PLC as described in Figure 14–8. H1receptors, the most prominent, are located on glia and vessels as well as on neurons. H3receptors, which have the greatest sensitivity to histamine, are localized primarily in basal ganglia and olfactory regions in rat brain. H4receptors are expressed on cells of hematopoietic origin: eosinophils, T cells, mast cells, basophils, and dendritic cells. H4 receptors are postulated to play a role in inflammation and chemotaxis. For details, consult Chapter 32.

Figure 14–8 Main signaling pathways for histamine receptors. Histamine can couple to a variety of G protein-linked signal transduction pathways via 4 different receptors. The H1 receptor and some H4receptors activate phosphatidylinositol turnover via Gq/11. The other receptors couple either positively (H2 receptor) or negatively (H3 and H4 receptor) to adenylyl cyclase activity via Gs and Gi/o.
Histaminergic neurons are located in the ventral posterior hypothalamus; they give rise to long ascending and descending tracts that are typical of the patterns characteristic of other aminergic systems. The histaminergic system is thought to affect arousal, body temperature, and vascular dynamics.
PEPTIDES. A remarkable array of neurotransmitters and neuromodulators have been described (Table 14–6). While some CNS peptides (see Table 14–6) may function on their own, most are now thought to act primarily in concert with coexisting transmitters (biogenic amines and amino acids), but their release can be independently regulated (see Figure 14–3). Many neuropeptides act via GPCRs (Table 14–7).
Table 14–6
Examples of Neuropeptides

Table 14–7
Peptide Transmitters and Receptors

Peptide synthesis takes place primarily in perikarya and the resulting peptide is then transported to nerve terminals. Single genes can, through posttranslational modifications, give rise to multiple neuropeptides. For example, proteolytic processing of proopiomelanocortin (POMC) gives rise to ACTH, α-, γ-, and β-MSH, and β-endorphin (Figure 14–9). In addition, alternative splicing of RNA transcripts may result in distinct mRNA species (e.g., calcitonin and calcitonin gene-related peptide [CGRP]).

Figure 14–9 Proteolytic processing of proopiomelanocortin (POMC). After removal of the signal peptide from pre-POMC, the remaining propeptide undergoes endoproteolysis by prohormone convertases 1 and 2 (PC1 and PC2) at dibasic residues. PC1 liberates the bioactive peptides adrenocorticotropic hormone (ACTH), t-endorphin (β end), and γ-lipotrophic hormone (γ-LPH). PC2 cleaves ACTH into corticotrophin-like intermediate lobe peptide (CLIP) and c-melanocyte stimulating hormone (α-MSH) and also releases γ-MSH from the N-terminal portion of the propeptide. The joining peptide (JP) is the region between ACTH and γ-MSH. β-MSH is formed by cleavage of γ-LPH. Some of the resulting peptides are amidated or acetylated before they become fully active.
CANNABINOIDS. Delta-9-tetrahydrocannabinol (THC) is 1 of several active substances in marijuana (Figure 14–10). The primary pharmacologic effects of THC follow its interaction with CB1receptors in the CNS and CB2receptors in the periphery. Both CB1 and CB2 receptors are linked to Gi and thence to inhibition of adenylyl cyclase activity. The natural endogenous ligands for these receptors are arachidonic acid derivatives including anandamide and 2-arachidonyl glycerol (see Figure 14–10).

Figure 14–10 Cannabinoid receptor ligands. Anandamide and 2-arachidonylglycerol are endogenous agonists. Rimonabant is a synthetic CB receptor antagonist. @del9-tetrahydrocannabinol is a CB agonist derived from marijuana.
THC has dramatic short-term effects, including causing feelings of euphoria and altered sensory perception. CB1 receptors are found primarily in the basal ganglia, hippocampus, cerebellum, and cerebral cortex; activation of CB1 receptors results in inhibition of glutamate release. Some nonneuronal cells and tissues also express CB1 receptors, including leukocytes and testis. CB2 receptors are expressed in the spleen, tonsils, bone marrow, and on peripheral blood leukocytes.
Efforts to develop CB1 antagonists like rimonabant (see Figure 14–10) have focused on possible treatments for drug addiction and obesity. Efforts are also underway to develop agonists that interact with CB1 and CB2 receptors for the relief of pain. THC (dronabinol [MARINOL]) is sometimes used in the control of nausea and moderate pain (see Chapter 46).
PURINES. Adenosine, ATP, UDP, and UTP have roles as extracellular signaling molecules. ATP is a component of the adrenergic storage vesicles and is released along with catecholamines. Intracellular nucleotides may also reach the cell surface by other means, and extracellular adenosine can result from cellular release and metabolism of ATP.
Extracellular nucleotides and adenosine act on a family of purinergic receptors that is divided into 2 classes, P1 and P2 (Table 14–8). P1 receptors are GPCRs that interact with adenosine; 2 of these receptors (A1 and A3) couple to Gi and 2 (A2a and A2b) couple to Gs; methylxanthines antagonize A1 and A3 receptors. Activation of A1 receptors is associated with inhibition of adenylyl cyclase, activation of K+ currents, and in some instances, with activation of PLC; stimulation of A2 receptors activates adenylyl cyclase. The P2 class includes a large number of P2X receptors that are ligand-gated ion channels and the P2Y receptors, a large subclass of GPCRs that couple to Gq or Gi. The P2Y14 receptor is expressed in the CNS; it interacts with UDP-glucose and may couple to Gq. The P2Y12 receptor is important clinically: inhibition of this receptor in platelets inhibits platelet aggregation.
Table 14–8
Characteristics of Purinergic Receptors

Although many of these receptors have been detected in brain, most of the current interest stems from pharmacological rather than physiological observations. Adenosine can act presynaptically throughout the cortex and hippocampal formation to inhibit the release of amine and amino acid transmitters. ATP-regulated responses have been linked pharmacologically to a variety of pathophysiological functions, including anxiety, stroke, and epilepsy. A2 receptors and dopamine D2 receptors appear to be functionally antagonistic, leading to investigation of A2a antagonists as adjunctive therapy for Parkinson disease.
LIPID MEDIATORS. Arachidonic acid, normally stored within the cell membrane as a glycerol ester, can be liberated during phospholipid hydrolysis (by pathways involving phospholipases A2, C, and D). Arachidonic acid can be converted to highly reactive regulators by 3 major enzymatic pathways (see Chapter 33): cyclooxygenases (leading to prostaglandins and thromboxane), lipoxygenases (leading to the leukotrienes and other transient catabolites of eicosatetraenoic acid), and CYPs (which are inducible and also expressed at low levels in brain). Arachidonic acid metabolites have been implicated as diffusible modulators in the CNS, possibly involved with the formation of LTP and other forms of neuronal plasticity.
NITRIC OXIDE (NO) AND CARBON MONOXIDE (CO). Both constitutive and inducible forms of NOS are expressed in the brain. The application of inhibitors of NOS (e.g., methylarginine) and of NO donors (such as nitroprusside) suggests the involvement of NO in a host of CNS phenomena, including LTP, activation of soluble guanylyl cyclase, neurotransmitter release, and enhancement of glutamate (NMDA)-mediated neurotoxicity. CO, generated in neurons, is another diffusable gas that may act as intracellular messenger stimulating soluble guanylyl cyclase.
CYTOKINES. Cytokines are a family of polypeptide regulators. The effects of cytokines are regulated by the conditions imposed by other cytokines, interacting as a network with variable effects leading to synergistic, additive, or opposing actions. Tissue-produced peptides, termed chemokines, serve to attract immune and inflammatory cells into interstitial spaces. These special cytokines have received attention as potential regulators of nervous system inflammation (as in early stages of dementia, following infection with human immunodeficiency virus, and during recovery from traumatic injury). Neurons and astrocytes may be induced under some pathophysiological conditions to express cytokines or other growth factors.
ACTIONS OF DRUGS IN THE CNS
SPECIFICITY AND NONSPECIFICITY OF CNS DRUG ACTIONS. The effect of a drug in the CNS is termed specific when it affects an identifiable molecular mechanism unique to target cells that bear receptors for that drug. Even a drug that is highly specific when tested at low concentrations may exhibit nonspecific actions at higher doses. In general, the more potent a drug at its desired target, the lower the probability that it will have off-target effects. Conversely, even drugs that have a broad spectrum of activity may not act identically at all levels of the CNS. For example, sedatives, hypnotics, and general anesthetics would have very limited utility if central neurons that control the respiratory and cardiovascular systems were especially sensitive to their actions. The specificity of a drug’s action is frequently overestimated. This is partly due to the fact that drugs are often identified with the effect that is implied by the class name.
GENERAL (NONSPECIFIC) CNS DEPRESSANTS. This category includes the anesthetic gases and vapors, the aliphatic alcohols, and some hypnotic-sedative drugs. These agents share the capacity to depress excitable tissue at all levels of the CNS, leading to a decrease in the amount of transmitter released by the nerve impulse, as well as to general depression of postsynaptic responsiveness and ion flux. At sub-anesthetic concentrations, these agents (e.g., ethanol) can exert relatively specific effects on certain groups of neurons, which may account for differences in their behavioral effects, especially the propensity to produce dependence.
GENERAL (NONSPECIFIC) CNS STIMULANTS. This category includes pentylenetetrazol and related agents that are capable of powerful excitation of the CNS, and the methylxanthines, which have a much weaker stimulant action. Stimulation may be accomplished by 1 of 2 general mechanisms: (1) blockade of inhibition or (2) direct neuronal excitation that may involve increased transmitter release or more prolonged transmitter action, as occurs when the reuptake of a released transmitter is inhibited.
DRUGS THAT SELECTIVELY MODIFY CNS FUNCTION. The agents in this group may cause either depression or excitation. In some instances, a drug may produce both effects simultaneously on different systems. The principal classes of these CNS drugs are anticonvulsants, drugs used in treating Parkinson disease, opioid and nonopioid analgesics, appetite suppressants, antiemetics, analgesic-antipyretics, certain stimulants, antidepressants, antimanic and antipsychotic agents, tranquilizers, sedatives and hypnotics, and medications employed in the treatment of Alzheimer disease (cholinesterase inhibitors and anti-glutamate neuroprotectants). Even if selectivity of action is remarkable, a drug usually affects several CNS functions to varying degrees.
GENERAL CHARACTERISTICS OF CNS DRUGS. Combinations of centrally acting drugs frequently are administered to therapeutic advantage (e.g., an anticholinergic drug and levodopa for Parkinson disease). However, other combinations of drugs may be detrimental because of potentially dangerous additive or mutually antagonistic effects. The effects of a CNS drug may be additive with the physiological state and the effects of other depressant and stimulant drugs. For example, anesthetics are less effective in a hyperexcitable subject than in a normal patient; the converse is true for stimulants. In general, the depressant effects of drugs from different categories are additive (e.g., the potentially fatal combination of barbiturates or benzodiazepines with ethanol), as are the effects of stimulants. Therefore, respiration depressed by morphine is further impaired by depressant drugs, while stimulant drugs can augment the excitatory effects of morphine to produce vomiting and convulsions.
Antagonism between depressants and stimulants is variable. Some instances of true pharmacological antagonism among CNS drugs are known; for example, opioid antagonists can selectively antagonize the effects of opioid analgesics. However, the antagonism exhibited between 2 CNS drugs is most often physiological in nature. For example, an individual whose CNS is depressed by an opiate cannot be returned entirely to normal by stimulation with caffeine.
The selective effects of drugs on specific neurotransmitter systems may be additive or competitive. The potential for drug interactions must be considered whenever such drugs are concurrently administered. To minimize such interactions, a drug-free period may be required when modifying therapy; in fact, development of desensitized and supersensitive states with prolonged therapy may limit the speed with which 1 drug may be halted and another started. An excitatory effect is commonly observed with low concentrations of certain depressant drugs due either to depression of inhibitory systems or to a transient increase in the release of excitatory transmitters. Examples include the stage of excitement seen during induction of general anesthesia. The excitatory phase typically occurs with low concentrations of the depressant; uniform depression ensues with increasing drug concentration. The excitatory effects can be minimized, when appropriate, by pretreatment with a depressant drug that is devoid of such effects (e.g., benzodiazepines in preanesthetic medication). Acute, excessive stimulation of the cerebrospinal axis normally is followed by depression, which is in part a consequence of neuronal fatigue and exhaustion of stores of transmitters. Postictal depression is additive with the effects of depressant drugs. Acute, drug-induced depression generally is not followed by stimulation. However, chronic drug-induced sedation or depression may be followed by prolonged hyperexcitability on abrupt withdrawal of the medication (barbiturates or alcohol). This type of hyperexcitability can be controlled effectively by the same or another depressant drug (see Chapters 17, 23, and 24).