Many new agents are available to block the fundamental mutations that cause specific cancers: aberrant growth factor receptors, dysregulated intracellular signaling pathways, defective DNA repair and apoptosis, and tumor angiogenesis. The primary tools for inhibiting these targets are monoclonal antibodies that attack cell surface receptors and antigens, and synthetic small molecules that enter cells and engage critical enzymes. These 2 classes of drugs have very different pharmacological properties.
Monoclonal antibodies kill tumor cells by blocking cell surface receptor function and by recruiting immune cells and complement to the antigen–antibody complex. They may be armed to carry toxins or radionuclides to the cells of interest, thereby enhancing their cytotoxic effects. They generally are specific for a single receptor, have a long plasma t1/2, and require only intermittent administration. Small molecules may attack the same targets and pathways as the monoclonals, but may also exert their effect by entering cells and inhibiting enzymatic functions (usually tyrosine kinase reactions). The small molecules often inhibit multiple enzymatic sites, have a broad spectrum of target kinases, and tend to be substrates of hepatic CYPs with a t1/2 of 12-24 h, and thus require daily oral administration.
These 2 drug classes, when targeted against the same pathway, may have significantly different spectra of antitumor activity. Thus, monoclonal antibodies to the epidermal growth factor receptor (EGFR) are effective in the treatment of head and neck and colon cancers, while small molecules, such as erlotinib and gefitinib, attack the intracellular tyrosine kinase function of the same receptor and have a different spectrum of antitumor activity (non–small cell lung cancer). The specific drug target is of central importance in cancer chemotherapy and forms the organizational basis for the discussion below.
PROTEIN TYROSINE KINASE INHIBITORS
There are 3 basic types of protein kinases (see Chapter 3):
• Kinases that specifically phosphorylate tyrosine residues
• Kinases that phosphorylate serine and threonine residues
• Kinases with activity toward all 3 residues
Tyrosine kinases can be further subdivided into those with an extracellular ligand-binding domain (receptor tyrosine kinases, associated with growth factor receptors, Figure 62–1) and intracellular enzymes (nonreceptor tyrosine kinases, e.g., src, abl, jak, fak, srm). In a number of human malignancies, mutations that constitutively activate protein tyrosine kinases are implicated in malignant transformation.
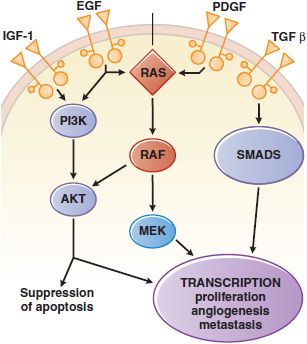
Figure 62–1 Growth factor signaling. Binding of agonist ligands to growth factor receptors (monospanning membrane proteins) causes receptor dimerization and activation of cytosolic protein kinase domains, leading to activation of multiple signaling pathways. Shown here are the RAS/MAPK/ERK, PI3K, and SMAD pathways, each of which is activated by receptors or cross-talk from adjacent pathways. Their signals regulate proliferation, metabolism, survival, and the synthesis of other growth factors, such as the vascular endothelial growth factor (VEGF).
INHIBITORS OF THE BCR-ABL KINASE: IMATINIB, DASATINIB, AND NILOTINIB
Imatinib mesylate (STI 571, GLEEVEC, GLIVEC) targets the BCR-ABL tyrosine kinase, which underlies chronic myelogenous leukemia (CML). A single molecular event, in this case the 9:22 translocation, leads to expression of the Abelson proto-oncogene kinase ABL fused to BCR (breakpoint cluster region), yielding a constitutively activated protein kinase, BCR-ABL, and then the malignant phenotype.
Imatinib and the related compounds dasatinib and nilotinib induce clinical and molecular remissions in >90% of CML patients in the chronic phase of disease. Imatinib treats other tumors that carry related tyrosine kinase mutations, including GI stromal tumors (driven by c-KIT mutation), and hypereosinophilia syndrome, chronic myelomonocytic leukemia, and dermatofibrosarcoma protuberans (all driven by mutations that activate the PDGF receptor [PDGFR]).
CHARACTERISTICS AND MECHANISM OF ACTION. Imatinib was identified through high-throughput screening against the BCR-ABL kinase. Dasatinib (BMS-354825, SPRYCEL), a second-generation BCR-ABL inhibitor, inhibits the Src kinase, and unlike imatinib, it binds both the open and closed configurations of the BCR-ABL kinase. Nilotinib (AMN107, TASIGNA) was designed to have increased potency and specificity compared to imatinib. Its structure overcomes mutations that cause imatinib resistance. Imatinib and nilotinib bind to a segment of the kinase domain that fixes the enzyme in a closed or nonfunctional state, in which the protein is unable to bind its substrate/phosphate donor, ATP. The 3 BCR-ABL kinase inhibitors differ in their potency of inhibition, their binding specificities, and their susceptibility to resistance mutations in the target enzyme. Dasatinib [(IC50)=<1 nM] and nilotinib [(IC50)=<20 nM] inhibit BCR-ABL kinase more potently than does imatinib [(IC50)=100 nM].
MECHANISMS OF RESISTANCE. Resistance to the tyrosine kinase inhibitors arises from point mutations in 3 separate segments of the kinase domain (see Figure 62–1 in the 12th edition of the parent text). The contact points between imatinib and the enzyme become sites of mutations in drug-resistant leukemic cells; these mutations prevent tight binding of the drug and lock the enzyme in its open configuration, in which it has access to substrate and is enzymatically active. Nilotinib retains inhibitory activity in the presence of most point mutations that confer resistance to imatinib. Other mutations affect the phosphate-binding region and the “activation loop” of the domain with varying degrees of associated resistance. Some mutations, such as at amino acids 351 and 355, confer low levels of resistance to imatinib, possibly explaining the clinical response of some resistant patients to dose escalation of imatinib.
Molecular studies have detected resistance-mediating kinase mutations prior to initiation of therapy, particularly in patients with Ph+ acute lymphoblastic leukemia (ALL) or CML in blastic crisis. This finding supports the hypothesis that drug-resistant cells arise through spontaneous mutation and expand under the selective pressure of drug exposure. Mechanisms other than BCR-ABL kinase mutation play a minor role in resistance to imatinib. Amplification of the wild-type kinase gene, leading to overexpression of the enzyme, has been identified in tumor samples from patients resistant to treatment. The multidrug resistant (MDR) gene confers resistance experimentally but has not been implicated in clinical resistance. Philadelphia chromosome-negative clones lacking the BCR-ABL translocation and displaying the karyotype of myelodysplastic cells may emerge in patients receiving imatinib for CML and may progress to myelodysplasia (MDS) and to acute myelocytic leukemia (AML). Their origin is unclear.
ADME
Imatinib. Imatinib is well absorbed after oral administration and reaches maximal plasma concentrations within 2-4 h. The elimination t1/2 of imatinib and its major active metabolite, the N-desmethyl derivative, are ~18 and 40 h, respectively. Food does not change the pharmacokinetic profile. Doses >300 mg/day achieve trough levels of 1 <M, which correspond to in vitro levels required to kill BCR-ABL–expressing cells. In the treatment of GI stromal cell tumors (GISTs), higher doses (600 mg/day) may improve response rates. CYP3A4 is the major metabolizer of imatinib; thus, drugs that induce or interact with CYP3A4 can alter the pharmacokinetics of imatinib. Coadministration of imatinib and rifampin, an inducer of CYP3A4, lowers the plasma imatinib AUC by 70%. Imatinib, as a competitive CYP3A4 substrate, inhibits the metabolism of simvastatin and increases its plasma AUC by 3.5-fold.
Dasatinib. Oral dasatinib is well absorbed; its bioavailability is significantly reduced at neutral gastric pH (i.e., after antacids and H2 blockers) but is unaffected by food. The plasmat1/2 of dasatinib is 3-5 h. Dasatinib exhibits dose proportional increases in AUC, and its clearance is constant over the dose range of 15-240 mg/day. Dasatinib doses of 70 mg twice a day, 100 mg daily, and 140 mg daily are equally effective in patients with CML, although the 100-mg daily dose improves progression-free survival. Dasatinib is metabolized primarily by CYP3A4, with minor contributions by FMO3 and UGT. Plasma concentrations of dasatinib are affected by inducers and inhibitors of CYP3A4 in a similar fashion to imatinib.
Nilotinib. Approximately 30% of an oral dose of nilotinib (400 mg 2 times daily) is absorbed after administration, with peak concentrations in plasma 3 h after dosing. Unlike the other BCR-ABL inhibitors, nilotinib’s bioavailability increases significantly in the presence of food. The drug has a plasma t1/2 17 h, and plasma concentrations reach a steady state only after 8 days of daily dosing. Nilotinib is metabolized by CYP3A4, with predictable alteration by inducers, inhibitors, and competitors of CYP3A4. Nilotinib is a substrate and inhibitor of P-glycoprotein.
THERAPEUTIC USES. These protein tyrosine kinase inhibitors have efficacy in diseases in which the ABL, kit, or PDGFR have dominant roles in driving the proliferation of the tumor, reflecting the presence of a mutation that results in constitutive activation of the kinase. Imatinib shows remarkable therapeutic benefits in patients with chronic-phase CML (BCR-ABL), GIST (kit mutation positive), chronic myelomonocytic leukemia (EVT6-PDGFR translocation), hypereosinophilia syndrome (FIP1L1-PDGFR), and dermatofibrosarcoma protuberans (constitutive production of the ligand for PDGFR). It is the agent of choice for GIST patients with metastatic disease and as adjuvant therapy of c-kit–positive GIST. The currently recommended dose of imatinib is 400-600 mg/day. Dasatinib is approved for patients with CML resistant or intolerant to imatinib in both chronic (100 mg/day) and advanced phases of disease (70 mg twice daily), and for use combined with cytotoxic chemotherapy in patients with Ph+ ALL who are resistant or intolerant to prior therapies. Nilotinib is approved for patients with CML resistant to or intolerant of prior imatinib therapy.
TOXICITY. Imatinib, dasatinib, and nilotinib cause GI symptoms (diarrhea, nausea, and vomiting) that are usually readily controlled. All 3 drugs promote fluid retention, edema, and peri-orbital swelling. Dasatinib may cause pleural effusions. Nilotinib may prolong the QT interval. Myelosuppression occurs infrequently but may require transfusion support, dose reduction, or discontinuation of the drug. These drugs can be associated with hepatotoxicity. Most nonhematological adverse reactions are self-limited and respond to dose adjustments. After the adverse reactions have resolved, the drug may be reinitiated and titrated back to effective doses.
EPIDERMAL GROWTH FACTOR RECEPTOR INHIBITORS
The EGFR belongs to the ErbB family of transmembrane receptor tyrosine kinases. EGFR, also known as ErbB1 or HER1, is essential for the growth and differentiation of epithelial cells. Ligand binding to the extracellular domain of EGFR family members causes receptor dimerization and stimulates the protein tyrosine kinase activity of the intracellular domain, resulting in autophosphorylation of several Tyr residues in the C-terminal domain. Recognition of the phosphotyrosines by other proteins initiates protein-protein interactions that result in stimulation of signaling pathways including MAPK, PI3K/Akt, and STAT pathways (see Figure 62–1). In epithelial cancers, overexpression and mutational activation of the EGFR are a common finding and create a dependence on EGFR signaling in these tumors.
Two separate classes of drugs that target the EGFR pathway are important agents in the therapy of solid tumors. The EGFR tyrosine kinase inhibitors erlotinib and gefitinib bind to the kinase domain and block the enzymatic function of EGFR. The monoclonal antibodies cetuximab and panitumumab bind specifically to the extracellular domain of EGFR and inhibit EGFR-dependent signaling.
GEFITINIB
Mechanism of Action. Gefitinib inhibits the EGFR tyrosine kinase by competitive blockade of ATP binding. Gefitinib has an IC50 of 20-80 nM for the EGFR tyrosine kinase but is significantly less potent against HER2 (ErbB2/neu). Gefitinib has antitumor activity in human xenograft tumors that exhibit high levels of EGFR expression.
ADME. Oral bioavailability is ~60%; peak plasma concentrations are achieved within 3-7 h. Absorption of gefitinib is not significantly altered by food but is reduced by drugs that cause elevations in gastric pH. Metabolism of gefitinib is predominantly via CYP3A4, with a terminal t1/2 of 41 h. Inducers of CYP3A4 activity decrease gefitinib plasma concentrations and efficacy; conversely, CYP3A4 inhibitors increase plasma concentrations.
Therapeutic Uses. Gefitinib initially was approved for the third-line treatment of patients with non–small cell lung cancer. However, a large clinical trial failed to show an effect on survival, leading the FDA to restrict its use to patients who have previously received clinical benefit from the drug. Two large trials have failed to demonstrate a benefit of gefitinib in combination with chemotherapy: patients who were nonsmokers, Asians, or women were most likely to respond to gefitinib. Tumors from these patients frequently have characteristic activating mutations in EGFR. The standard dose is 250 mg/day.
Adverse Effects and Drug Interactions. Diarrhea and pustular/papular rash occur in ~50% of patients. Other side effects include dry skin, nausea, vomiting, pruritus, anorexia, and fatigue. Most adverse effects occur within the first month of therapy and are manageable with medications and dose reductions. Asymptomatic increases in liver transaminases may necessitate dose reduction or discontinuation of therapy. Interstitial lung disease occurs in <2% of patients and may have a fatal outcome. Inducers and inhibitors of CYP3A4 will alter plasma concentrations. Patients using warfarin should be monitored closely for elevation of the international normalized ratio (INR) while taking gefitinib.
ERLOTINIB
Mechanism of Action. Erlotinib (TARCEVA) is a potent inhibitor of the EGFR tyrosine kinase, competitively inhibiting ATP binding at the active site of the kinase. Erlotinib has an IC50 of 2 nM for the EGFR kinase. Tumors harboring k-ras mutations and EML4-ALK translocations do not respond to EGFR kinase inhibitors.
ADME. Erlotinib is ~60% absorbed after oral administration but should not be taken with food, which increases its bioavailability to ~100%. Peak plasma levels occur after 4 h. Erlotinib has a t1/2 of 36 h and is metabolized by CYP3A4 and to a lesser extent by CYPs 1A2 and 1A1. The standard daily dose of erlotinib results in a plasma AUC ~10-fold greater than the AUC of gefitinib.
Therapeutic Uses. Erlotinib is approved for second-line treatment of patients with locally advanced or metastatic non–small cell lung cancer. Erlotinib also is approved for first-line treatment of patients with locally advanced, unresectable, or metastatic pancreatic cancer in combination with gemcitabine. The recommended dose of erlotinib in non–small cell lung cancer is a 150-mg tablet daily. In pancreatic cancer, the dose is a 100-mg tablet daily, taken at least 1 h before or 2 h after a meal.
Adverse Effects and Drug Interactions. The most common adverse reactions are diarrhea, an acneform rash, anorexia, and fatigue. Serious or fatal interstitial lung disease occurs with a frequency of 0.7-2.5%. Serious or fatal hepatic failure due to erlotinib has been reported, particularly in patients with baseline hepatic dysfunction. Other rare but serious toxicities include GI perforation, renal failure, arterial thrombosis, microangiopathic hemolytic anemia, hand-foot skin reaction, and corneal perforation or ulceration. Erlotinib therapy may cause rare cases of Stevens-Johnson syndrome/toxic epidermal necrolysis.
Concurrent use of proton-pump inhibitors decreases the bioavailability of erlotinib by 50%. Plasma levels can vary due to drug interactions with inducers or inhibitors of CYP3A4. Patients using warfarin may experience elevations of the INR while taking erlotinib. Smoking accelerates metabolic clearance of erlotinib and may decrease its antitumor effects.
RESISTANCE TO GEFITINIB AND ERLOTINIB
Patients with non–small cell lung cancer who initially respond to erlotinib or gefitinib have tumors that are dependent on the EGFR signaling pathway. Tumors containing mutations in EGFR initially respond to erlotinib and gefitinib but eventually progress. A secondary mutation in the EGFR gatekeeper residue, T790M, prevents binding of drug to the kinase domain and confers resistance. Other potential mechanisms of resistance include activation of downstream mediators, efflux of drug, and altered receptor trafficking. Therapy directed at the EGFR may delay disease progression in patients with non–small cell lung cancers that lack activating EGFR mutations, although response rates approach zero in these patients.
CETUXIMAB
Cetuximab (ERBITUX) is a recombinant chimeric human/mouse immunoglobulin G1 (IgG1) antibody that binds to the extracellular domain of EGFR. Such antibodies, although sharing the same target with erlotinib and gefitinib and having a similar side effect profile, have a different spectrum of antitumor activity.
Mechanism of Action. Cetuximab binds specifically to the extracellular domain of EGFR and prevents ligand-dependent signaling and receptor dimerization, thereby blocking cell growth and survival signals. Cetuximab also may mediate antibody-dependent cellular cytotoxicity against tumor cells.
ADME. Cetuximab exhibits nonlinear pharmacokinetics. Following intravenous administration, steady-state levels are achieved by the third weekly infusion. Therapeutic doses that saturate total body receptor pools of EGFR follow zero-order kinetics for elimination. Clearance occurs via EGFR binding and internalization and by degradation in the reticuloendothelial system.
Therapeutic Uses
Head and Neck Cancer. Cetuximab is used in combination with radiation therapy for locally or regionally advanced squamous cell carcinoma of the head and neck (HNSCC). It also is indicated in monotherapy for patients with metastatic or recurrent HNSCC who fail platinum-based chemotherapy. It is a useful agent in combination with cisplatin-based chemotherapy.
Metastatic Colon Cancer. Cetuximab is approved as a single agent for the treatment of EGFR-positive metastatic colorectal cancer; cetuximab is used in patients who cannot tolerate irinotecan-based therapy and in combination with irinotecan for patients refractory to oxaliplatin, irinotecan, and 5-fluorouracil (5-FU). In the first-line setting, cetuximab may improve survival in combination with 5-FU/leucovorin and irinotecan or oxaliplatin. About 40-50% of colorectal tumors carry mutations in the k-ras oncogene and are resistant to the effects of cetuximab. Cetuximab enhances the effectiveness of chemotherapy in patients with k-ras mutant tumors but not k-ras wild-type tumors. The standard dose of cetuximab is a single loading dose of 400 mg/m2 intravenously, followed by weekly doses of 250 mg/m2 intravenously for the duration of treatment.
Adverse Effects. Side effects include an acneform rash (in the majority of patients), pruritus, nail changes, headache, and diarrhea. Rare but serious adverse effects include cardiopulmonary arrest, interstitial lung disease, and hypomagnesemia. In addition, patients can develop anaphylactoid reactions during infusion, which may be related to preexisting IgE antibodies that are more prevalent in patients from the southern U.S.
PANITUMUMAB
Panitumumab (VECTIBIX) is a recombinant, fully humanized IgG2k antibody that binds specifically to the extracellular domain of EGFR. Unlike cetuximab, it does not mediate antibody-dependent cell-mediated cytotoxicity.
Panitumumab exhibits nonlinear pharmacokinetic characteristics. Following intravenous administration every 2 weeks, steady-state levels are achieved by the third infusion. The mean t1/2 is 7.5 days. Panitumumab improves progression-free survival in patients with metastatic colorectal carcinoma. The dose of panitumumab is 6 mg/kg intravenously given once every 2 weeks. The adverse effects with panitumumab are similar to cetuximab and include rash and dermatological toxicity, severe infusion reactions, pulmonary fibrosis, and electrolyte abnormalities.
HER2/NEU INHIBITORS
Both antibodies (trastuzumab, pertuzumab) and small molecules (lapatinib et al.) have striking antitumor effects in patients with HER2-positive breast cancer, and have become essential therapeutic agents in combination with cytotoxic chemotherapy for this aggressive malignancy.
TRASTUZUMAB. Trastuzumab (HERCEPTIN) is a humanized monoclonal antibody that binds to the external domain of HER2/neu (ErbB2).
Thirty percent of breast cancers overexpress this receptor due to gene amplification on chromosome 17. Amplification of the receptor is associated with lower response rates to hormonal therapies and to most cytotoxic drugs, with the exception of anthracyclines. Patients with HER2/neu-amplified tumors have higher recurrence rates after standard adjuvant therapy and poorer overall survival. The internal domain of the HER2/neu glycoprotein encodes a tyrosine kinase that activates downstream signal, enhances metastatic potential, and inhibits apoptosis. Trastuzumab exerts its antitumor effects through: inhibition of homo- or heterodimerization of receptor, thereby preventing receptor kinase activation and downstream signaling; initiation of Fcγ-receptor-mediated antibody-dependent cellular cytotoxicity; and blockade of the angiogenetic effects of HER2 signaling.
Therapeutic Uses. Trastuzumab is approved for HER2/neu-overexpressing metastatic breast cancer, in combination with paclitaxel as initial treatment or as monotherapy following chemotherapy relapse. Trastuzumab synergizes with other cytotoxic agents in HER2/neu-overexpressing cancers.
Pharmacokinetics and Toxicity. Trastuzumab has dose-dependent pharmacokinetics with a mean t1/2 of 5.8 days on the 2-mg/kg maintenance dose. Steady-state levels are achieved between 16 and 32 weeks. The infusional effects of trastuzumab are typical for monoclonal antibodies and include fever, chills, nausea, dyspnea, and rashes. Premedication with diphenhydramine and acetaminophen is indicated. The most serious toxicity of trastuzumab is cardiac failure; reasons for cardiotoxicity are poorly understood. Before initializing therapy, baseline electrocardiogram and cardiac ejection fraction measurement should be obtained to rule out underlying heart disease, and patients deserve careful clinical follow-up thereafter for signs or symptoms of congestive heart failure. When trastuzumab is used as a single agent, <5% of patients will experience a decrease in left-ventricular ejection fraction, and 1% will have clinical signs of congestive failure. Left-ventricular dysfunction occurs in up to 20% of patients who receive the antibody in combination with doxorubicin and cyclophosphamide. The risk of cardiac toxicity is greatly reduced with taxane–trastuzumab combinations.
LAPATINIB. Lapatinib and other pan-HER inhibitors block both ErbB1 and ErbB2 and bind to an internal site on the receptor (usually the ATP-binding pocket), compared to the external binding site of trastuzumab. Lapatinib also inhibits a truncated form of the HER2 receptor that lacks a trastuzumab-binding domain, differences that may account for the activity of lapatinib in trastuzumab-resistant patients.
Therapeutic Uses. Lapatinib (TYKERB) is approved for HER2-amplified, trastuzumab-refractory breast cancer, in combination with the fluoropyrimidine analog, capecitabine. As a small molecule, lapatinib crosses the blood-brain barrier more readily than inhibitor antibodies and has produced anecdotal responses in patients with brain metastases in phase III trial.
Pharmacokinetics and Toxicity. The drug is administered orally, 1250 mg/day. It is metabolized by CYP3A4 to inactive products and oxidized to an intermediate that has activity against ErbB1 but not ErbB2. The plasma t1/2 is 14 h. Concurrent administration of inducers and inhibitors of CYP3A4 may necessitate adjustment of the dose. Lapatinib toxicities include mild diarrhea, cramping, and exacerbation of gastro-esophageal reflux. When lapatinib is combined with capecitabine, diarrhea becomes a significant side effect (33%). Lapatinib causes an acneform rash in one-third of patients that may be controlled with topical benzoyl peroxide gel. Lapatinib has no clear signal of cardiac toxicity; nonetheless, because it targets ErbB2, lapatinib should be used with caution in combination with other cardiotoxic drugs and with careful surveillance in patients with heart disease.
INHIBITORS OF ANGIOGENESIS
Cancer cells secrete angiogenic factors that induce the formation of new blood vessels and guarantee the flow of nutrients to the tumor cells. Angiogenic factors secreted by tumors include VEGF (vascular endothelial growth factor), FGF (fibroblast growth factor), TGF-β (transforming growth factor β), and PDGF (platelet-derived growth factor). Multiple tumor types overexpress these angiogenic factors. Tumor secretion of pro-angiogenic factors turns on an “angiogenic switch,” a process essential to tumor growth and metastasis. In multiple experimental models, blockade of these pro-angiogenic molecules halts tumor growth, and in human cancers, anti-angiogenic drugs also have inhibitory effects.
Leaky capillaries within tumors have increased permeability and cause an increase in tumor interstitial pressure that inhibits blood flow, decreases oxygenation, and prevents drug delivery within the tumor. Antibodies directed at the primary angiogenic factor, VEGF, “normalize” interstitial pressure, improve blood flow, and enhance the ability of chemotherapeutic agents to reach the tumor. Hence, an additional benefit of anti-angiogenic molecules may be their ability to increase the delivery of chemotherapy to the tumor. This hypothesis seems to be validated in the synergy observed when cytotoxic chemotherapy is combined with anti-VEGF antibodies.
VEGF initiates endothelial cell proliferation when it binds to a member of the VEGF receptor (VEGFR) family, a group of highly homologous receptors with intracellular tyrosine kinase domains that includes VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4). The binding of VEGF to its receptor activates the intracellular VEGFR tyrosine kinase activity and initiates mitogenic and anti-apoptotic signaling pathways. Antibodies targeting VEGF, such as bevacizumab, sterically hinder the interaction of VEGF with its receptor. Aflibercept (ZALITRAP), acts as a VEGF Trap; it is a recombinant molecule that uses the VEGFR1-binding domain to sequester VEGF and acts as a “soluble decoy receptor” for VEGF. Three small molecules (pazopanib, sorafenib, and sunitinib) that inhibit the kinase function of VEGFR-2 have been approved for clinical use. Although bevacizumab and the small molecules share a similar spectrum of toxicities, they have different spectra of clinical activity and pharmacokinetics.
BEVACIZUMAB
Bevacizumab (AVASTIN), a humanized antibody directed against VEGF-A, delays progression of renal-cell cancer, and, in combination with cytotoxic chemotherapy, effectively treats lung, colorectal, and breast cancers.
ADME. Bevacizumab is administered intravenously as a 30- to 90-min infusion. In metastatic colon cancer, in conjunction with combination chemotherapy, the dose of bevacizumab is 5 mg/kg every 2 weeks. In metastatic non–small cell lung cancer, doses of 15 mg/kg are given every 3 weeks with chemotherapy. For metastatic breast cancer, patients receive 10 mg/kg of bevacizumab every 2 weeks in combination with paclitaxel or docetaxel. The antibody has a plasma t1/2 of 4 weeks.
Therapeutic Uses. In clear-cell renal-cell carcinoma, a cancer notoriously resistant to traditional chemotherapeutic agents, single-agent bevacizumab increases survival by 3 months. Bevacizumab is approved in combination with interferon-α for the treatment of metastatic renal-cell carcinoma. Bevacizumab is approved as a single agent following prior therapy for glioblastoma. In all other cancers, bevacizumab has little apparent single-agent activity but improves survival in epithelial cancers in combination with standard chemotherapeutic agents. Bevacizumab with carboplatin and paclitaxel increases survival in non–small lung cancer by 2 months. Likewise, bevacizumab combined with FOLFOX (5-FU, leucovorin, and oxaliplatin) or FOLFIRI (5-FU, leucovorin, and irinotecan) improves survival by 5 months in metastatic colon cancer. Finally, the combination of bevacizumab with docetaxel increases progression-free survival in patients with metastatic breast cancer.
A slightly altered version of bevacizumab, ranibizumab (LUCENTIS), in which the Fc region has been deleted, effectively treats wet macular degeneration. Bevacizumab restores hearing in patients with progressive deafness due to neurofibromatosis type 2–related tumors.
Toxicity. Bevacizumab causes a wide range of class-related adverse effects. A prominent concern is the potential for vessel injury and bleeding in patients with squamous-cell lung cancer. Bevacizumab is contraindicated for patients who have a history of hemoptysis, brain metastasis, or a bleeding diathesis, but in appropriately selected patients, the rate of life-threatening pulmonary hemorrhage is <2%. The most dreaded vascular toxicity of anti-angiogenic agents is an arterial thromboembolic event (i.e., stroke or myocardial infarction). The rate of arterial thromboembolic events in patients receiving bevacizumab-containing regimens is 3.8% compared to the control rate of 1.7%. To reduce the risk of arterial thromboembolic events, clinicians should evaluate a patient’s risk factors (age >65 years, a past history of arterial thromboembolic events) before starting the drug.
Other toxicities characteristic of anti-angiogenic drugs include hypertension and proteinuria. A majority of patients receiving the drug require antihypertensive therapy, particularly those receiving higher doses and more prolonged treatment. Bevacizumab is rarely associated with congestive heart failure, probably secondary to hypertension, and with reversible posterior leukoencephalopathy in patients with poorly controlled hypertension. GI perforation, a potentially life-threatening complication, has been observed (up to 11%) in patients with ovarian cancer. In colon cancer patients, colonic perforation occurs infrequently during bevacizumab treatment but increases in frequency in patients with intact primary colonic tumors, peritoneal carcinomatosis, peptic ulcer disease, chemotherapy-associated colitis, diverticulitis, or prior abdominal radiation treatment. The rate of colon perforation is <1% in breast and lung cancer patients. Colon cancer surgery patients on bevacizumab have a higher rate (13% vs. 3.4%) of serious wound healing complications. Because of the long t1/2 of bevacizumab, elective surgery should be delayed for at least 4 weeks from the last dose of antibody, and treatment should be not resumed for at least 4 weeks after surgery.
SUNITINIB
Sunitinib (SUTENT) competitively inhibits the binding of ATP to the tyrosine kinase domain on the VEGF receptor-2, a mechanism it shares with sorafenib. Sunitinib also inhibits other protein tyrosine kinases at concentrations of 5-100 nM.
Pharmacokinetics. Sunitinib is administered orally in doses of 50 mg once a day. The typical cycle of sunitinib is 4 weeks on treatment followed by 2 weeks off. The dosage and schedule of sunitinib can be increased or decreased according to toxicity (hypertension, fatigue). Dosages <25 mg/day typically are ineffective. Sunitinib is metabolized by CYP3A4 to produce an active metabolite, SU12662, the t1/2 of which is 80-110 h; steady-state levels of the metabolite are reached after ~2 weeks of repeated administration of the parent drug. Further metabolism results in the formation of inactive products. The pharmacokinetics of sunitinib are not affected by food intake.
Therapeutic Uses. Sunitinib has activity in metastatic renal-cell cancer, producing a higher response rate (31%) and a longer progression-free survival than any other approved anti-angiogenic drug. Sunitinib also is approved for treatment of advanced renal-cell carcinoma, GIST that have developed resistance to imatinib as a consequence of c-KIT mutations, and pancreatic neuroendocrine tumors. Specific c-KIT mutations correlate with degree of response to sunitinib (e.g., patients with c-KIT exon 9 mutations have a response rate of 37%; patients with c-KIT exon 11 mutations have only a 5% response rate).
Toxicity. The main toxicities of sunitinib are shared by all anti-angiogenic inhibitors: bleeding, hypertension, proteinuria, and, uncommonly, arterial thromboembolic events and intestinal perforation. However, because sunitinib is a multi-targeted tyrosine kinase inhibitor, it has a broader side-effect profile than bevacizumab. Fatigue affects 50-70% of patients and may be disabling. Hypothyroidism occurs in 40-60% of patients. Bone marrow suppression and diarrhea also are common side effects; severe neutropenia (neutrophils <1000/mL) develop in 10% of patients. Less common side effects include congestive heart failure (usually in association with hypertension) and hand-foot syndrome. It is essential to check blood counts and thyroid function at regular intervals. Periodic echocardiograms also are recommended.
SORAFENIB
Sorafenib (NEXAVAR), like sunitinib, targets multiple protein tyrosine kinases and inhibits their catalytic activities at concentrations of 20-90 nM. Sorafenib is the only drug currently approved for treatment of hepatocellular carcinoma. Sorafenib also is approved in metastatic renal-cell cancer, for which sunitinib generally is the preferred first-line therapy.
ADME. Sorafenib is given orally, beginning with 200 mg once a day and increasing to 400 mg twice a day as tolerated. Sorafenib is given every day without treatment breaks. Sorafenib is metabolized to inactive products by CYP3A4 with a t1/2 of 20-27 h; with repeated administration, steady-state concentrations are reached within 1 week.
Adverse Effects. Sorafenib patients can experience the vascular toxicities seen with other anti-angiogenic medications. More common adverse effects include fatigue, nausea, diarrhea, anorexia, and rash; uncommonly, bone marrow suppression and GI perforation.
IMMUNOMODULATORS (IMiDs)
Among agents with anti-angiogenic activity, the immunomodulatory analogs (IMiDs) thalidomide and lenalidomide have a most unusual history and a multiplicity of biological and immunological effects. Thalidomide originally was used for the treatment of pregnancy-associated morning sickness but was withdrawn from the market due to teratogenicity and dysmelia (stunted limb growth). It re-entered clinical practice for treatment of erythema nodosum leprosum (see Chapter 56). Further research revealed its anti-angiogenic and immunomodulatory effects. At least 4 distinct mechanisms have been proposed to explain the antitumor activity of IMiDs as summarized by Figure 62–2 and enumerated in its legend.
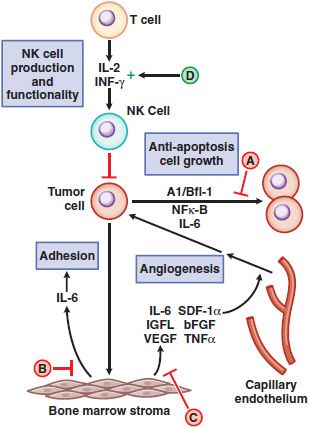
Figure 62–2 Schematic overview of proposed mechanisms of antimyeloma activity of thalidomide and its derivatives. Some biological hallmarks of the malignant phenotype are indicated in blue boxes. The proposed sites of action for thalidomide (letters inside red and green circles) are hypothesized to also be operative for thalidomide derivatives. A. Direct anti–multiple myeloma (MM) effect on tumor cells, including G1 growth arrest and/or apoptosis, even against MM cells resistant to conventional therapy. This is due to the disruption of the anti-apoptotic effect of BCL-2 family members, blocking NF-κB signaling, and inhibition of the production of interleukin-6 (IL-6). B. Inhibition of MM-cell adhesion to bone marrow stromal cells partially due to the reduction of IL-6 release. C. Decreased angiogenesis due to the inhibition of cytokine and growth factor production and release. D. Enhanced T-cell production of cytokines, such as IL-2 and interferon-γ (IFN-γ), that increase the number and cytotoxic functionality of natural killer (NK) cells. VEGF, vascular endothelial growth factor.
Both thalidomide and lenalidomide possess potent activity in newly diagnosed and heavily pretreated relapsed/refractory multiple myeloma (MM) patients. Lenalidomide also is approved for its activity in the 5q–subset of MDS. A specific gene array profile identifies MDS patients who lack the 5q–abnormality but respond to lenalidomide.
THALIDOMIDE
ADME. Thalidomide (THALOMID) exists at physiological pH as a racemic mixture of cell-permeable and rapidly interconverting non-polar S(–) and R(+) isomers. The R-enantiomer is associated with the teratogenic and biological activities, while the S accounts for the sedative properties of thalidomide. Thalidomide is given in dosages of 200-1200 mg/day. In treating MM, doses usually are escalated by 200 mg/day every 2 weeks until dose-limiting side effects (sedation, fatigue, constipation, or a sensory neuropathy) supervene. With extended treatment, the neuropathy may necessitate dose reduction or discontinuation of treatment for a period of time. Thalidomide absorption from the GI tract is slow and highly variable. It distributes throughout most tissues and organs, without significant binding to plasma proteins. The enantiomers are eliminated with a t1/2 ≈6 h, mainly due to spontaneous hydrolysis in all body fluids; the S-enantiomer is cleared more rapidly than the R-enantiomer. Thalidomide and its metabolites are excreted in the urine, while the non-absorbed portion of the drug is excreted unchanged in feces. The inactive hydrolysis products undergo CYP-mediated metabolism. Longer plasma t1/2is reported at highest doses (1200 mg daily). No dose adjustment is necessary in the presence of renal failure.
Thalidomide enhances the sedative effects of barbiturates and alcohol and the catatonic effects of chlorpromazine and reserpine. Conversely, CNS stimulants (such as methamphetamine and methylphenidate) counteract the depressant effects of thalidomide.
LENALIDOMIDE
Lenalidomide (REVLIMID) constitutes the lead compound of immunomodulatory thalidomide derivatives. Pharmacological properties include: direct suppression of the tumor cell growth in culture, T-cell and NK-cell activation, suppression of TNF-α and other cytokines, anti-angiogenesis, and promotion of hematopoietic stem cell differentiation.
ADME. The standard dosage of lenalidomide is 25 mg/day for 21 days of a 28-day cycle. The drug is rapidly absorbed following oral administration, reaching peak plasma levels within 1.5 h. The t1/2 of parent drug in plasma is 9 h. Approximately 70% of the orally administered dose of lenalidomide is excreted intact by the kidney. Dose adjustments to 10 mg/day for creatinine clearance of 30-50 mL/h and to the same dose every 2 days for creatinine clearance <30 mL/h are recommended for patients with renal failure.
Therapeutic Use. Lenalidomide exhibits potent antitumor activity in MM, MDS, and chronic lymphocytic leukemia (CLL); this agent causes fewer adverse side effects and lacks the teratogenicity of thalidomide.
ADVERSE EFFECTS OF THALIDOMIDE AND LENALIDOMIDE
Thalidomide is well tolerated at doses <200 mg daily. Common adverse effects are sedation and constipation; the most serious one is peripheral sensory neuropathy, which occurs in 10-30% of patients with MM or other malignancies in a dose- and time-dependent manner. Thalidomide-related neuropathy is an asymmetrical, painful, peripheral paresthesia with sensory loss, commonly presenting with numbness of toes and feet, muscle cramps, weakness, signs of pyramidal tract involvement, and carpal tunnel syndrome. Although symptoms improve upon drug discontinuation, long-standing sensory loss may not reverse. Particular caution should apply in patients with pre-existing neuropathy (e.g., related to diabetes) or prior exposure to drugs that can cause peripheral neuropathy (e.g., vinca alkaloids, bortezomib).
Adverse effects of lenalidomide are much less severe; it causes little sedation, constipation, or neuropathy. The drug depresses bone marrow function and is associated with significant leukopenia (20% of patients). Hepatotoxicity and renal dysfunction are rare. In some CLL patients, lenalidomide causes dramatic lymph node swelling and tumor lysis (tumor flare reaction). Patients with renal dysfunction are prone to this reaction; thus, CLL patients should be started at lower doses of 10 mg/day, with escalation as tolerated. CLL patients should receive pretreatment hydration and allopurinol to avoid the consequences of tumor swelling and tumor lysis. A negative interaction with rituximab, an anti-CD20 antibody, may result from lenalidomide’s downregulation of CD20, an interaction that has clinical implications for their combined use in lymphoid malignancies.
Thromboembolic events occur with increased frequency in patients receiving thalidomide or lenalidomide, but particularly in combination with glucocorticoids and with anthracyclines. Anticoagulation reduces this risk and seems indicated in patients presenting with risk factors for clotting.
PROTEASOME INHIBITION: BORTEZOMIB
Bortezomib (VELCADE), an inhibitor of proteasome-mediated protein degradation, has a central role in the treatment of MM. Bortezomib has a unique boron-containing structure:
Mechanism of Action. Bortezomib binds to the β5 subunit of the 20S core of the 26S proteasome and reversibly inhibits its chymotrypsin-like activity. This event disrupts multiple intracellular signaling cascades, leading to apoptosis. An important consequence of proteasome inhibition is its effect on NF-κB, a transcription factor that promotes cell damage response and cell survival. Most cellular NF-κB is cytosolic and bound to IBB; in this form, NF-κB is restricted to the cytosol and cannot enter to the nucleus to regulate transcription. In response to stress signals resulting from hypoxia, chemotherapy, and DNA damage, IBB becomes ubiquitinated and then degraded via the proteasome. Its degradation releases NF-κB, which enters the nucleus and transcriptionally activates a host of genes involved in cell survival (e.g., cell adhesion proteins E-selectin, ICAM-1, and VCAM-1), as proliferation (e.g., cyclin-D1) or anti-apoptosis (e.g., cIAPs, BCL-2). NF-κB is highly expressed in many human tumors, including MM, and may be a key factor in tumor cell survival in a hypoxic environment and during chemotherapy. Bortezomib blocks proteasomal degradation of IkB, thereby preventing the transcriptional activity of NF-κB and downregulating survival responses.
Bortezomib also disrupts the ubiquitin-proteasomal degradation of p21, p27, p53, and other key regulators of the cell cycle and initiators of apoptosis. Bortezomib activates the cell’s stereotypical “unfolded protein response” or UPR, in which abnormal protein conformation activates adaptive signaling pathways in the cell. The composite effect leads to irreversible commitment of MM cells to apoptosis. Bortezomib also sensitizes tumor cells to cytotoxic drugs, including alkylators and anthracyclines, and to IMiDs and inhibitors of histone deacetylase.
ADME. The recommended starting dose of bortezomib is 1.3 mg/m2 given as an intravenous bolus on days 1, 4, 8, and 11 of every 21-day cycle (with a 10-day rest period per cycle). At least 72 h should elapse between doses. Drug administration should be withheld until resolution of any grade 3 nonhematological toxicity or grade 4 hematological toxicity, and subsequent doses should be reduced 25%. The drug exhibits a terminal t1/2 in plasma of 5.5 h. Peak proteasome inhibition reaches 60% within 1 h and declines thereafter, with a t1/2 of ~24 h. Bortezomib clearance results from the deboronation of the parent compound (90%), followed by hydroxylation of the boron-free product by CYPs 3A4 and 2D6; administration of this drug with potent inducers or inhibitors/substrates of CYP3A4 requires caution. No dose adjustment is required for patients with renal dysfunction.
Therapeutic Uses. Bortezomib is used as initial therapy for MM and as therapy for MM after relapse from other drugs. It also is approved for relapsed or refractory mantle cell lymphoma. The drug is active in myeloma, including the induction of complete responses in up to 30% of patients when used in combination with other drugs (i.e., thalidomide, lenalidomide, liposomal doxorubicin, or dexamethasone).
Toxicity. Bortezomib toxicities include thrombocytopenia (28%), fatigue (12%), peripheral neuropathy (12%), and neutropenia, anemia, vomiting, diarrhea, limb pain, dehydration, nausea, or weakness. Peripheral neuropathy, the most chronic of the toxicities, develops most frequently in patients with a prior history of neuropathy secondary to prior drug treatment (e.g., thalidomide) or diabetes or with prolonged use. Dose reductions or discontinuation of bortezomib ameliorates the neuropathic symptoms. Injection of bortezomib may precipitate hypotension, especially in volume-depleted patients, in those who have a history of syncope, or in patients taking antihypertensive medications. Cardiac toxicity is rare, but congestive failure and prolonged QT-interval have been reported.
mTOR INHIBITORS: RAPAMYCIN ANALOGS
Rapamycin (sirolimus) is a fungal fermentation product that inhibits serine/threonine protein kinase in mammalian cells eponymously named mammalian target of rapamycin, or mTOR. The PI3K/PKB(Akt)/mTOR pathway responds to a variety of signals from growth factors. The activation of the PI3K pathway is opposed by the phosphatase activity of the tumor suppressor, PTEN. Activating mutations and amplification of genes in the receptor-PI3K pathway, and loss of function alterations in PTEN, occur frequently in cancer cells, with the result that PI3K signaling is exaggerated and cells exhibit enhanced survival.
Rapamycin and its congeners, temsirolimus and everolimus, are first-line drugs in post-transplant immunosuppression (see Figure 35–1). The mTOR inhibitors have important applications in oncology for treatment of renal and hepatocellular cancer and mantle cell lymphomas. Everolimus was recently FDA-approved for the treatment of advanced pancreatic neuroendocrine tumors.
Mechanisms of Action and Resistance. The rapamycins inhibit an enzyme complex, mTORC1, which occupies a downstream position in the PI3 kinase pathway (Figure 62–3). mTOR forms the mTORC1 complex with a member of the FK506-binding protein family, FKBP12. Among other actions, mTORC1 phosphorylates S6 kinase and also relieves the inhibitory effect of 4EBP on initiation factor eiF-4E, thereby promoting protein synthesis and metabolism. The antitumor actions of the rapamycins result from their binding to FKBP12 and inhibition of mTORC1. Rapamycin and its congeners have immunosuppressant effects, inhibit cell-cycle progression and angiogenesis, and promote apoptosis. Resistance to mTOR inhibitors is incompletely understood but may arise through the action of a second mTOR complex, mTORC2, which is unaffected by rapamycins and which regulates Akt.
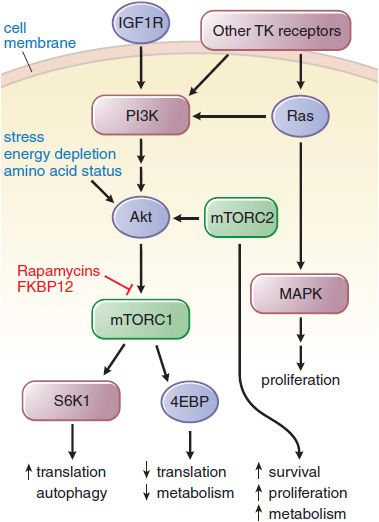
Figure 62–3 Effect of rapamycin on growth factor signaling. A key pathway is regulated by phosphatidylinositol-3 kinase (PI3K) and its downstream partner, the mammalian target of rapamycin (mTOR). Rapamycins complex with FKBPP12 to inhibit the mTORC1 complex. mTORC2 remains unaffected and responds by upregulating Akt, driving signals through the inhibited mTORC1. The various downstream outputs of the 2 complexes are shown. Phosphorylation of 4EBP by mTOR inhibits the capacity of 4EBP to inhibit eif-4E and slow metabolism. 4EBP, eukaryotic initiation factor 4e (eif-4E) binding protein; FKBP12, immunophilin target (binding protein) for tacrolimus (FK506); IGF1R, insulin-like growth factor 1 receptor; S6K1, S6 kinase 1.
ADME. For renal-cell cancer, temsirolimus is given in weekly doses of 25 mg, intravenously; everolimus is administered orally in doses of 10 mg/day. Both drugs should be administered in the fasting state at least 1 h before a meal. Both parent molecules are metabolized by CYP3A4. Temsirolimus has a plasma t1/2 of 30 h; its primary metabolite, sirolimus, has a t1/2 of 53 h. Because sirolimus has equivalent activity as an inhibitor of mTORC1, and has a greater AUC, sirolimus is likely the more important contributor to antitumor action in patients. Everolimus has a plasma t1/2 of 30 h; on a weekly schedule at doses of 20 mg, it maintains inhibition of mTORC1 for 7 days in white blood cells. Both drugs are susceptible to interactions with other agents that affect CYP3A4 activity. The dose of temsirolimus should be doubled in the presence of inducers and reduced by half in the presence of ketoconazole. For everolimus, the dose should be reduced to 5 mg daily for patients with moderate hepatic impairment (Child-Pugh class B); guidelines for dose reduction of temsirolimus in such patients have not been established. The drugs’ pharmacokinetics do not depend on renal function, and hemodialysis does not hasten temsirolimus clearance.
Therapeutic Uses. Temsirolimus and everolimus are approved for treatment of renal cancer. Temsirolimus prolongs survival and delays disease progression in patients with advanced and poor- or intermediate-risk renal cancer, as compared to standard interferon-α treatment. Everolimus, as compared to placebo, prolongs survival in patients who had failed initial treatment with anti-angiogenic drugs. mTOR inhibitors also have antitumor activity against mantle cell lymphomas.
Toxicity. The rapamycin analogs have very similar patterns of toxicity. Side effects such as a mild maculopapular rash, mucositis, anemia, and fatigue occur in 30-50% of patients. A few patients will develop leukopenia or thrombocytopenia, effects that are reversed if therapy is discontinued. Less common side effects include hyperglycemia, hypertriglyceridemia, and, rarely, pulmonary infiltrates and interstitial lung disease. Pulmonary infiltrates emerge in 8% of patients receiving everolimus and in a smaller percentage of those treated with temsirolimus. If symptoms such as cough or shortness of breath develop or radiological changes progress, the drug should be discontinued. Prednisone may hasten the resolution of radiological changes and symptoms.
BIOLOGICAL RESPONSE MODIFIERS
Biological response modifiers include cytokines and monoclonal antibodies that beneficially affect the patient’s biological response to a neoplasm. Such agents may act indirectly to mediate their antitumor effects (e.g., by enhancing the immunological response to neoplastic cells) or directly, binding to receptors on the tumor cells and delivering toxins or radionuclides. Proteins that currently are in clinical use include the interferons, interleukins, hematopoietic growth factors (e.g., erythropoietin, filgrastim [granulocyte colony-stimulating factor], and sargramostim [granulocyte-macrophage colony-stimulating factor]; see Chapter 37), and monoclonal antibodies.
MONOCLONAL ANTIBODIES
Cancer cells express antigens that are attractive targets for monoclonal antibody–based therapy (Table 62–1). Immunization of mice with human tumor cell extracts has led to the isolation of monoclonal Abs reactive against unique or highly expressed target antigens, and a few of these monoclonals possess antitumor activity. Because murine antibodies have a short t1/2 in humans, activate human immune effector mechanisms poorly, and induce a human anti-mouse antibody immune response, they usually are chimerized by substituting major portions of the human IgG molecule. The nomenclature adopted for naming therapeutic monoclonal antibodies is to terminate the name in -ximab for chimeric antibodies and -umab for fully humanized antibodies.
Table 62–1
Monoclonal Antibodies Approved For Hematopoietic And Solid Tumors
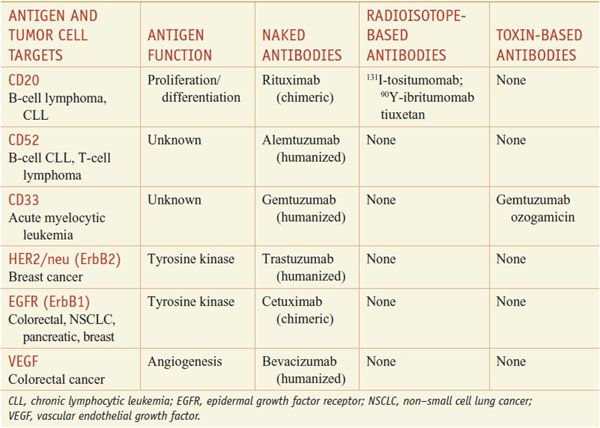
Presently, monoclonal antibodies are FDA-approved for lymphoid and solid tumor malignancies. Available agents include rituximab and alemtuzumab for lymphoid malignancies, trastuzumab for breast cancer, bevacizumab for colon and lung cancer, and cetuximab and panitumumab for colorectal cancer and head and neck cancer. Table 62–1 summarizes available monoclonal antibodies and their tumor cell targets; Table 62–2 summarizes the mechanisms, dose regimens, and toxicities of monoclonal antibody-based agents. Unmodified monoclonal antibodies may kill tumor cells by a variety of mechanisms (e.g., antibody-dependent cellular cytotoxicity [ADCC], complement-dependent cytotoxicity [CDC], and direct induction of apoptosis by antigen binding), but the clinically relevant mechanisms for most antibodies are uncertain. Monoclonal antibodies also may be linked to a toxin (immunotoxins), such as gemtuzumab ozogamicin (MYLOTARG) or denileukin diftitox (ONTAK), or conjugated to a radioactive isotope, as in the case of 90Yttrium (90Y)-ibritumomab tiuxetan (ZEVALIN) (see Table 62–1).
UNARMED MONOCLONAL ANTIBODIES
RITUXIMAB. Rituximab (RITUXAN) is a chimeric monoclonal antibody that targets the CD20 B-cell antigen (Tables 62–1 and 62–2).
Table 62–2
Mechanism, Dose Regimen, and Toxicity Monoclonal Antibody-Based Drugs
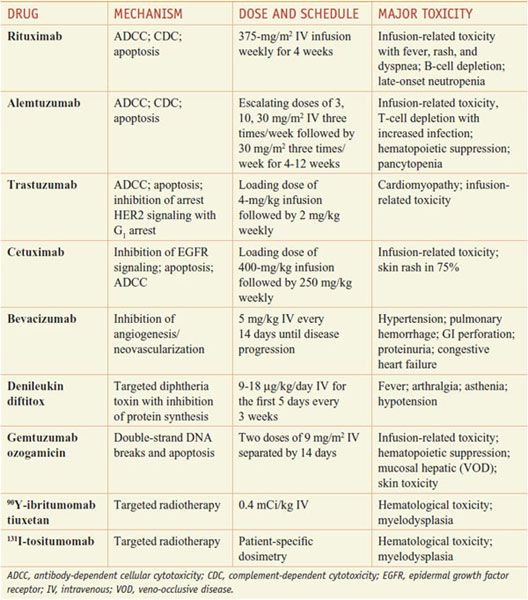
CD20 is found on cells from the pre–B cell stage through its terminal differentiation to plasma cells and is expressed on 90% of B-cell neoplasms. Monoclonal antibody binding to CD20 generates transmembrane signals that produce autophosphorylation and activation of protein serine/tyrosine kinases, induction of c-myc oncogene expression, and expression of major histocompatibility complex class II molecules. CD20 also may regulate transmembrane Ca2+ conductance through its function as a Ca2+ channel. It is unclear which of these actions relates to the pharmacological effects of rituximab.
ADME. Rituximab has a t1/2 of ~22 days. The drug is administered by intravenous infusion both as a single agent and in combination with chemotherapy at a dose of 375 mg/m2. As a single agent, it is given weekly for 4 weeks, with maintenance dosing every 3-6 months. In combination regimens, the drug may be administered every 3-4 weeks, with chemotherapy, for up to 8 doses. As maintenance therapy following 6-8 cycles of combination chemotherapy, rituximab may be given once weekly for 4 doses, at 6-month intervals, for up to 16 doses. The rate of infusion should be increased slowly to prevent serious hypersensitivity reactions. Infusions should begin at 50 mg/h, and in the absence of infusion reactions, the rate can increase in 50-mg/h increments every 30 min to a maximum rate of 400 mg/h. Pretreatment with antihistamines, acetaminophen, and glucocorticoids decreases the risk of hypersensitivity reactions. Patients with large numbers of circulating tumor cells (as in CLL) are at increased risk for tumor lysis syndrome; in these patients, the initial dose should be no more than 50 mg/m2 on day 1 of treatment, and patients should receive standard tumor lysis prophylaxis. The remainder of the dose can then be given on day 3.
Therapeutic Uses. Rituximab is approved as a single agent for relapsed indolent lymphomas and significantly enhances response and survival in combination with chemotherapy for the initial treatment of diffuse large B-cell lymphoma. Rituximab improves response rates when added to combination chemotherapy for other indolent B-cell non-Hodgkin lymphomas (NHLs), including CLL, mantle cell lymphoma, Waldenström macroglobulinemia, and marginal zone lymphomas. Maintenance of remission with rituximab delays time to progression and improves overall survival in indolent NHL. It is increasingly used for treatment of autoimmune diseases such as rheumatological disease, thrombotic thrombocytopenic purpura, autoimmune hemolytic anemias, cryoglobulin-induced renal disease, and multiple sclerosis.
Resistance and Toxicity. Resistance to rituximab may emerge through downregulation of CD20, impaired antibody-dependent cellular cytotoxicity, decreased complement activation, limited effects on signaling and induction of apoptosis, and inadequate blood levels. Polymorphisms in 2 of the receptors for the antibody Fc region responsible for complement activation may predict the clinical response to rituximab monotherapy in patients with follicular lymphoma but not in CLL. Rituximab infusional reactions can be life-threatening, but with pretreatment are usually mild and limited to fever, chills, throat itching, urticaria, and mild hypotension. All respond to decreased infusion rates and antihistamines. Uncommonly, patients may develop severe mucocutaneous skin reactions, including Stevens-Johnson syndrome. Rituximab may cause reactivation of hepatitis B virus or rarely, JC virus (with progressive multifocal leukoencephalopathy). Hypogammaglobulinemia and autoimmune syndromes (idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, autoimmune hemolytic anemia, pure red cell aplasia, and delayed neutropenia) may supervene 1-5 months after administration (see Table 62–2).
OFATUMUMAB
Ofatumumab (ARZERRA) is a second monoclonal antibody that binds to CD20 at sites on the major and minor extracellular loops of CD20, distinct from the site targeted by rituximab. Binding of the drug results in B-cell lysis (both CDC and ADCC). Ofatumumab is approved for treating CLL after failure of fludarabine and alemtuzumab. A complex dosing scheme is used, beginning with small (300 mg) doses on day 1, followed by higher doses (up to 2 g/week) for 7 weeks, followed by 2 g every 4 weeks for 4 additional doses. Ofatumumab’s primary toxicities consist of immunosuppression and opportunistic infection, hypersensitivity reactions during antibody infusion, and myelosuppression. Blood counts should be monitored during treatment. Rarely, patients may develop reactivation of viral infections. The drug should not be administered to patients with active hepatitis B infection; liver function should be monitored in hepatitis B carriers.
ALEMTUZUMAB
Alemtuzumab (CAMPATH) is a humanized IgG-κ) monoclonal antibody. The drug binds to CD52 antigen present on the surface of a subset of normal neutrophils and on all B and T lymphocytes, on testicular elements and sperm, and on most B- and T-cell lymphomas. Consistently high levels of CD52 expression on lymphoid tumor cells and the lack of CD52 modulation with antibody binding make this antigen a favorable target for unconjugated monoclonal antibodies. Alemtuzumab can induce tumor cell death through ADCC and CDC.
ADME; Therapeutic Uses. Alemtuzumab is administered intravenously in dosages of 30 mg/day, 3 times/week. Premedication with diphenhydramine (50 mg) and acetaminophen (650 mg) should precede drug infusion. Dosing should begin with a 3-mg infusion, followed by a 10-mg dose 2 days later and, if well tolerated, a 30-mg dose 2 days later. The drug has an initial mean t1/2 of 1 h, but after multiple doses, the t1/2 extends to 12 days, and steady-state plasma levels are reached at approximately week 6 of treatment, presumably through saturation of CD52-binding sites. Clinical activity has been demonstrated in both B- and T-cell low-grade lymphomas and CLL, including patients with disease refractory to purine analogs. In chemotherapy-refractory CLL, overall response rates are ~40%, with complete responses of 6% in multiple series. Response rates in patients with untreated CLL are higher (overall response rates of 83% and complete responses of 24%).
Toxicity. Serious toxicities include acute infusion reactions and depletion of normal neutrophils and T cells (see Table 62–2). Myelosuppression, with depletion of all blood lineages, occurs in the majority of patients and may represent either direct marrow toxicity or auto-immune responses. Immunosuppression leads to a significant risk of fungal, viral, and other opportunistic infections, particularly in patients who have previously received purine analogs. Patients should receive antibiotic prophylaxis against Pneumocystis carinii and herpes virus during treatment and for at least 2 months following therapy with alemtuzumab. Because reactivation of cytomegalovirus (CMV) infections may follow antibody use, patients should be monitored for symptoms and signs of viremia, hepatitis, and pneumonia. CD4+ T-cell counts may remain profoundly depleted (<200 cells/µL) for 1 year. Alemtuzumab does not combine well with chemotherapy in standard regimens because of significant infectious complications.
IPILIMUMAB
Ipilimumab (YERVOY) is a human monoclonal antibody against CTLA-4, approved for the treatment of late stage inoperable melanoma. The agent potentiates T cell proliferation by removing inhibition by cytotoxic T lymphocyte antigen-4 (CTLA-4). Ipilimumab treatment is associated with severe and fatal immunological adverse effects.
MONOCLONAL ANTIBODY–CYTOTOXIN CONJUGATES
GEMTUZUMAB OZOGAMICIN
Gemtuzumab ozogamicin (MYLOTARG) is a humanized monoclonal antibody against CD33 covalently linked to a semisynthetic derivative of calicheamicin, a potent antitumor antibiotic. The CD33 antigen is present on most hematopoietic cells, on >80% of AMLs, and on most myeloid cells in patients with myelodysplasias. However, other normal cell types lack CD33 expression, making this antigen attractive for targeted therapy. CD33 has no known biological function, although monoclonal antibody cross-linking inhibits normal and myeloid leukemia cell proliferation. Following its binding to CD33, gemtuzumab ozogamicin undergoes endocytosis; cleavage of calicheamicin from the antibody takes place within the lysosome. The potent toxin then enters the nucleus, binds in the minor groove of DNA, and causes double-strand DNA breaks and cell death.
Clinical Pharmacology. The antibody conjugate produces a 30% complete response rate in relapsed AML, when administered at a dose of 9 mg/m2 for up to 3 doses at 2-week intervals. The t1/2 of total and unconjugated calicheamicin are 41 h and 143 h, respectively. Following a second dose, the t1/2 of drug-antibody conjugate increases to 64 h. Most patients require 2 to 3 doses to achieve remission. The drug is approved in patients >60 years of age with AML in first relapse. Primary toxicities include myelosuppression in all patients treated and hepatocellular damage in 30-40% of patients, manifested by hyperbilirubinemia and enzyme elevations. It also causes a syndrome that resembles hepatic veno-occlusive disease when patients subsequently undergo myeloablative therapy or when gemtuzumab ozogamicin follows high-dose chemotherapy. Defibrotide, an orphan drug, may prevent severe or fatal hepatic injury in patients who develop signs of hepatic failure while receiving a stem cell transplant following gemtuzumab ozogamicin. Prolonged myelosuppression may complicate the patient’s course following remission induction with gemtuzumab ozogamicin.
RADIOIMMUNOCONJUGATES
Radioimmunoconjugates provide targeted delivery of radionuclides to tumor cells (Tables 62–1 and 62–2). 131Iodine (131I) is a favored radioisotope because it is readily available, relatively inexpensive, and easily conjugated to a monoclonal antibody. The γ particles emitted by 131I can be used for both imaging and therapy, but protein-iodine conjugates have the drawback of releasing free 131I and 131I-tyrosine into the blood, and thus present a health hazard to people in contact with the patient. The β-emitter 90Y has emerged as an alternative to 131I, based on its higher energy and longer path length. Thus, it may be more effective in tumors with larger diameters. It also has a short t1/2 and remains conjugated, even after endocytosis, providing a safer profile for outpatient use.
Currently available radioimmunoconjugates consist of murine monoclonal antibodies against CD20 conjugated with 131I (tositumomab [BeXXAR]) or 90Y (ibritumomab tiuxetan [ZEVALIN]). Both drugs have shown responses rates in relapsed lymphoma of 65-80%. When using Zevalin, an initial dose of unlabeled rituximab is administered, followed by an imaging dose of Indium-111-labeled Zevalin. Biodistribution is determined, allowing calculation of a therapeutic dose of 90Y Zevalin. A pretreatment dose of rituximab then is administered to saturate nonspecific binding sites, followed by the therapeutic dose of 90Y Zevalin. The steps in Bexxar administration closely follow those of Zevalin. An imaging dose precedes the therapeutic dose given 1 week later. These agents cause antibody-related hypersensitivity, bone marrow suppression, and secondary leukemias.
INTERLEUKIN-2
IL-2 is a 133–amino acid glycoprotein (MW ~ 15 kDa) produced by activated T cells and NK cells; it promotes activated T-cell proliferation and enhanced killing by NK cells. Responsiveness depends on expression of the IL-2 receptor. Resting T cells and nearly all types of tumor cells lack receptor expression and are unresponsive to IL-2.
The IL-2 receptor has 3 components: (1) an α chain, a 55-kDa protein (CD25) involved mainly in IL-2 binding; (2) a β chain, a 75-kDa protein involved in intracellular signaling; and (3) a γ chain, a 64-kDa protein that is a component of many cytokine receptors (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21) and also is involved in signaling. In the absence of the α chain, IL-2-binding affinity is reduced by a factor of 100.
Mechanism of Action. IL-2 stimulates the proliferation of activated T cells and the secretion of cytokines from NK cells and monocytes. IL-2 stimulation increases cytotoxic killing by T cells and NK cells. The mechanism of tumor cell killing has not been precisely defined.
Pharmacokinetics. The serum t1/2 of IL-2 after intravenous administration has an α phase of about 13 min and a β phase of about 90 min. IL-2 is excreted in the urine as an inactive metabolite.
Therapeutic Use. Aldesleukin (PROLEUKIN) possesses the biological activities of human native IL-2. The drug is approved for use in metastatic renal-cell cancer and metastatic melanoma. High-dose IL-2 produces an overall response rate of ~19% in patients with renal-cell cancer, and 8% achieve a complete response. Responses last a median of 8-9 years. High-dose IL-2 induces an overall response rate of ~16% in patients with metastatic melanoma, and 6% achieve a complete response. Responses last a median of ~5 years. Low-dose IL-2 also produces responses, but the duration of the responses may be less than with high-dose IL-2. Aldesleukin may be administered in several ways. High-dose IL-2 is 600,000-720,000 IU/kg administered by intravenous bolus every 8 h until dose-limiting toxicity appears or 14 total doses have been given; the schedule may be repeated after 9 days of rest for a maximum of 28 doses. Low-dose IL-2 is 60,000 or 72,000 IU/kg given by intravenous bolus every 8 h for 15 doses. A third regimen involves delivery of 18 million units/m2 daily by continuous intravenous infusion for 5 days. Chronic administration doses are 250,000 IU/kg subcutaneously daily for 5 days followed by 125,000 IU/kg daily for 6 weeks.
Toxicity. IL-2 toxicities are dominated by the capillary leak syndrome in which intravascular fluid leaks into the extravascular space, producing hypotension, edema, respiratory difficulties, confusion, tachycardia, oliguric renal failure, and electrolyte problems. Symptoms include fever, chills, malaise, nausea, vomiting, and diarrhea. Laboratory abnormalities include thrombocytopenia, abnormal liver function tests, and neutropenia. Most patients develop a pruritic skin rash. Hypothyroidism may occur. Arrhythmias are a rare complication. These toxicities can be life-threatening, yet nearly all are reversible within 24-48 h of discontinuing therapy. Patients should have normal renal and hepatic function before beginning therapy and should be closely supervised during drug administration.
DENILEUKIN DIFTITOX
Denileukin diftitox (ONTAK) is an immunotoxin made from the genetic recombination of IL-2 and the catalytically active fragment of diphtheria toxin. The high-affinity IL-2R has limited tissue expression and is an attractive target for an immunotoxin. Introduction of the diphtheria toxin fragment into cells leads to ADP-ribosylation and inactivation of eukaryotic elongation factor EF-2, inhibition of protein synthesis, and thence, cell death.
Clinical Pharmacology. Denileukin diftitox is approved for the treatment of recurrent/refractory cutaneous T-cell lymphomas. It should be administered at 9 or 18 µg/kg/day by intravenous infusion over 30-60 min for 5 consecutive days every 21 days for 8 cycles. Response rates of 30-37% have been achieved, with a median response duration of 6.9 months. The drug is quickly distributed and has a terminal t1/2 of ~70 min. Denileukin diftitox clearance in later cycles of treatment accelerates by 2- to 3-fold as a result of development of antibodies, but serum levels exceed those required to produce cell death in IL-2R-expressing cell lines (1-10 ng/mL for >90 min). Patients with a history of hypersensitivity reactions to diphtheria toxin or IL-2 should not be treated. Significant toxicities include acute hypersensitivity reactions, a vascular leak syndrome, and constitutional toxicities; glucocorticoid premedication significantly decreases toxicity.
COLONY-STIMULATING FACTORS
Many agents used for cancer chemotherapy suppress the production of multiple types of hematopoietic cells, and bone marrow suppression can limit the delivery of chemotherapy on schedule and at prescribed doses. The availability of recombinant growth factors for erythrocytes (i.e., erythropoietin), granulocytes (i.e., G-CSF), and granulocytes and macrophages (i.e., GM-CSF) has advanced the ability to use combination therapy or high-dose therapy with diminished complications such as febrile neutropenia (see Chapter 37).
NEWLY APPROVED FIRST-IN-CLASS DRUGS
Vemurafenib (ZELBORAF) is an orally administered selective inhibitor of the BRAF serine-threonine kinase that is FDA-approved for the treatment of metastatic melanoma harboring activating BRAF (V600E) mutations.
Crizotinib (XALKORI) is an ATP-competitive oral small-molecule inhibitor of the anaplastic lymphoma kinase (ALK) and mesenchymal-epithelial transition factor (c-MET) tyrosine kinase. The FDA granted accelerated approval to crizotinib for the treatment of patients with locally advanced or metastatic non–small cell lung cancer harboring ALK gene rearrangements.
Omacetaxine mepesuccinate (SYNRIBO) injection is an alkaloid that inhibits protein translation by preventing initial elongation in protein synthesis. It is approved for treatment of adults with chronic myeloid leukemia (CML) and with resistance to two or more tyrosine kinase inhibitors.
Vismodegib (ERIVEDGE) is a first-in-class hedgehog signaling pathway inhibitor approved for the treatment of adult patients with basal cell carcinoma. It acts as a competitive antagonist of the SMO receptor preventing the activation of the GLI family transcription factors.
For details of other approvals from 2011 to the present, see updates by Dr. Nelda Murri on the G&G webpages at AccessMedicine.com and AccessPharmacy.com.