In the steady state, Na+ intake via the gastrointestinal tract equals Na+ output from renal and extrarenal pathways
The two principal solutes in the ECF are Na+ and Cl−. Sodium is one of the most abundant ions in the body, totaling ~58 meq/kg body weight. Approximately 65% of the total Na+ is located in the ECF, and an additional 5% to 10%, in the intracellular fluid (ICF). Extracellular and intracellular Na+, comprising 70% to 75% of the total-body pool, is readily exchangeable, as determined by its ability to equilibrate rapidly with injected radioactive Na+. The remaining 25% to 30% of the body's Na+ pool is bound as Na+-apatites in bone. The concentration of Na+ in the plasma and interstitial fluid typically ranges between 135 and 145 mM.
Chloride totals ~33 meq/kg body weight. Approximately 85% is extracellular, and the remaining 15% is intracellular. Thus, all Cl− is readily exchangeable. The [Cl−] of plasma and interstitial fluid normally varies between 100 and 108 mM. Changes in total-body Cl− are usually influenced by the same factors, and in the same direction, as changes in total-body Na+. Exceptions arise during acid-base disturbances, when Cl− metabolism may change independently of Na+.
By definition, in the steady state, the total-body content of water and electrolytes is constant. For Na+, this concept can be expressed as
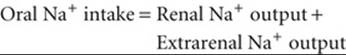
(40-1)
Under normal circumstances, extrarenal Na+ output is negligible. However, large fluid losses from the gastrointestinal tract (e.g., vomiting, diarrhea) or skin (e.g., excessive sweating, extensive burns) can represent substantial extrarenal Na+ losses. The kidney responds to such deficits by reducing renal Na+ excretion. Conversely, in conditions of excessive Na+ intake, the kidneys excrete the surfeit of Na+.
The kidneys increase Na+ excretion in response to an increase in ECF volume, not to an increase in extracellular Na+ concentration
In contrast to many other renal mechanisms of electrolyte excretion, the renal excretion of Na+ depends on the amount of Na+ in the body and not on the Na+ concentration in ECF. Because the amount of Na+ is the product of ECF volume and the extracellular Na+ concentration, and because the osmoregulatory system keeps plasma osmolality constant within very narrow limits, it is actually the volume of ECF that acts as the signal for Na+ homeostasis.
Figure 40-1A demonstrates the renal response to an abrupt step increase and step decrease in Na+ intake. A subject weighing 70 kg starts with an unusually low Na+ intake of 10 mmol/day, matched by an equally low urinary output. When the individual abruptly increases dietary Na+ intake from 10 to 150 mmol/day—and maintains it at this level for several days—urinary Na+ output also increases, but at first it lags behind intake. This initial period during which Na+ intake exceeds Na+ output is a state of positive Na+ balance. After ~5 days, urinary Na+ output rises to match dietary intake, after which total-body Na+does not increase further. In this example, we assume that the cumulative retention of Na+ amounts to 140 mmol.
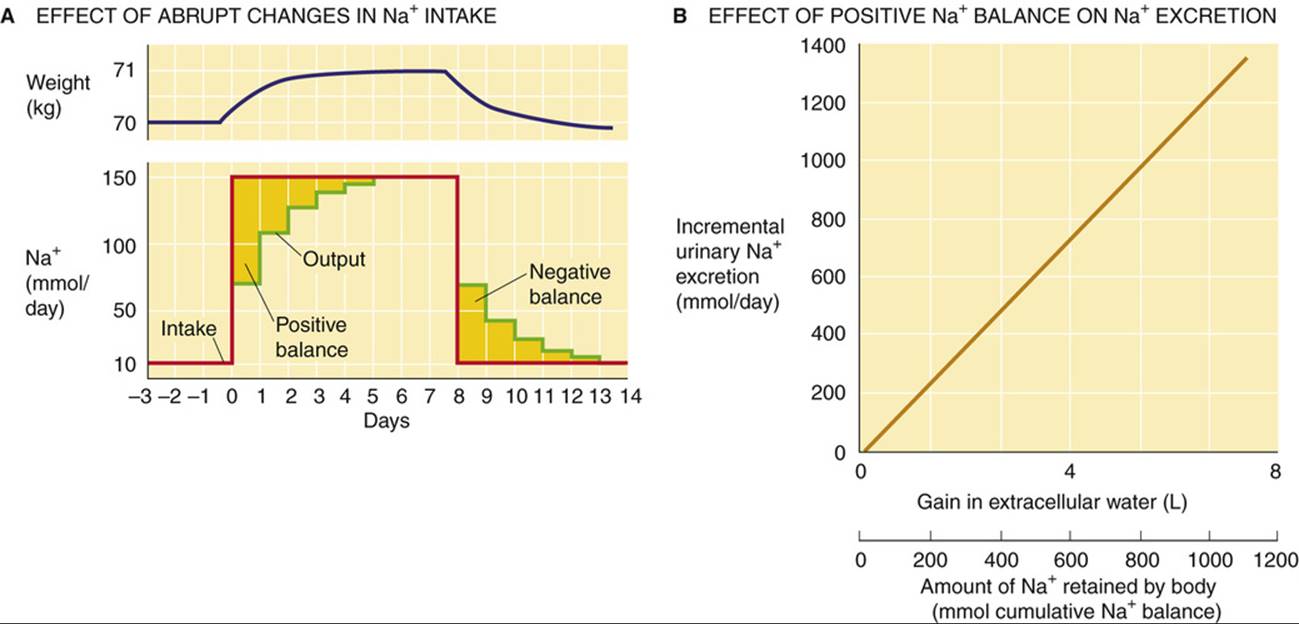
FIGURE 40-1 Na+ balance. In A, the red curve shows the time course of dietary Na+ intake, and the green curve shows Na+ excretion. The gold area between the two curves at the beginning of the experiment corresponds to the accumulated total-body Na+ of 140 mmol. This additional Na+, dissolved in ~1 L of ECF, accounts for the 1-kg gain in body weight (blue curve). (B, Data from Walser M: Phenomenological analysis of renal regulation of sodium and potassium balance. Kidney Int 27:837–841, 1985.)
The abrupt increase in dietary Na+ initially elevates plasma osmolality, thus stimulating thirst and release of AVP. Because the subject has free access to water, and because the kidneys salvage water in response to AVP (see pp. 817–819), the volume of free water rises. This increase in free water not only prevents a rise in [Na+] and osmolality, but also produces a weight gain that, in this example, is 1 kg (see Fig. 40-1A). This weight gain corresponds, in our example, to the accumulation of 140 mmol of Na+ and the accompanying free water, which makes 1 L of isotonic saline. In the new steady state, only the extracellular compartment has increased in volume. Intracellular volume does not change because, in the end, no driving force exists for water to cross cell membranes (i.e., extracellular osmolality is normal). Instead, the slight expansion of ECF volume signals the kidney to increase its rate of Na+ excretion. The extracellular Na+ concentration is unchanged during this period and thus cannot be the signal to increase Na+ excretion.
When the subject in our example later reduces Na+ intake to the initial level of 10 mmol/day (see Fig. 40-1A), Na+ excretion diminishes until the initial balanced state (input = output) is established once again. Immediately after the reduction in Na+ intake, Na+ is temporarily out of balance. This time, we have a period of negative Na+ balance, in which output exceeds input. During this period, the ECF volume falls by 1 L, and body weight returns to normal. Again, the extracellular Na+ concentration is unchanged during this transient period.
Ingestion of increasingly larger amounts of Na+ results in retention of progressively larger amounts in the steady state and thus accumulation of progressively more ECF volume. Urinary Na+ excretion increases linearly with this rise in retained Na+, as shown in Figure 40-1B. The control system that so tightly links urinary Na+ excretion to ECF volume is extremely sensitive. In our hypothetical example (see Fig. 40-1A)—a 70-kg individual with an initial ECF volume of 17 L—expanding ECF volume by 1 L, or ~6%, triggers a 15-fold increase in steady-state urinary Na+ excretion (i.e., from 10 mmol/day to 150 mmol/day in Fig. 40-1A). Physiologically normal individuals can be in Na+ balance on a nearly Na+-free diet (1 to 2 mmol/day) without overt signs of ECF volume depletion. Conversely, even with consumption of a high-Na+ diet (200 mmol/day versus the “normal” ~100 mmol/day for a Western diet), clinical signs of ECF volume excess, such as edema, are absent.
It is not the ECF volume as a whole, but the effective circulating volume, that regulates Na+ excretion
Although we have referred to the overall expansion of the ECF volume as the signal for increased urinary Na+ excretion, this is an oversimplification. Only certain regions of the ECF compartment are important for this signaling. For an expansion in ECF volume to stimulate Na+ excretion—either acutely or chronically—the expansion must make itself evident in parts of the ECF compartment where the ECF volume sensors are located, namely, in blood-filled compartments. ECF volume per se is not the critical factor in regulating renal Na+ excretion.
The critical parameter that the body recognizes is the effective circulating volume (see pp. 554–555)—not something that we can identify anatomically. Rather, effective circulating volume is a functional blood volume that reflects the extent of tissue perfusion in specific regions, as evidenced by the fullness or pressure within their blood vessels. Normally, changes in effective circulating volume parallel those in total ECF volume. However, this relationship may be distorted in certain diseases, such as congestive heart failure, nephrotic syndrome, or liver cirrhosis. In all three cases, total ECF volume is grossly expanded (e.g., edema or ascites). In contrast, the effective circulating volume is low, resulting in Na+ retention. For example, in congestive heart failure, particularly when edema is extensive, the total ECF volume is greatly increased. However, the low cardiac output fails to expand the key blood-filled compartments. As a result, Na+ reabsorption by the renal tubules remains high (i.e., urinary Na+ excretion is inappropriately low compared with Na+ intake), which exacerbates the systemic congestion (Box 40-1). ![]() N40-1
N40-1
Box 40-1
Volume Expansion and Contraction
When Na+ intake persists in the face of impaired renal Na+ excretion (e.g., during renal failure), the body retains isosmotic fluid. The result is an expansion of plasma volume and of the interstitial fluid compartment. In the extreme, the interstitial volume increase can become so severe that the subepidermal tissues swell (e.g., around the ankles). When the physician presses with a finger against the skin and then removes the finger, the finger imprint remains in the tissue—pitting edema. Not all cases of lower-extremity edema reflect total-body Na+ and fluid retention. For example, venous obstruction to return of blood from the lower extremities can cause local edema in the lower legs. Patients with this condition should elevate their feet whenever possible and should wear compression stockings.
Fluid can also accumulate in certain transcellular spaces (see p. 102), such as the pleural cavity (pleural effusion) or the peritoneal cavity (ascites); such conditions reflect derangements of local Starling forces or an increase in protein permeability due to inflammation, which alters the fluid distribution between the plasma and the ECF (see Box 20-1). In cases of abnormal Na+ retention, putting the subject on a low-Na+ diet can partially correct the edema. Administration of diuretics ![]() N40-2 can also reduce volume overload, as long as the kidney retains sufficient function to respond to them.
N40-2 can also reduce volume overload, as long as the kidney retains sufficient function to respond to them.
An excessive loss of Na+ into the urine can be caused by disturbances of Na+ reabsorption along the nephron and leads to a dramatic shrinkage of the ECF volume. Because the plasma volume is part of the ECF volume, significant reductions can severely affect the circulation, culminating in hypovolemic shock (see p. 583). Renal causes of reduced ECF volume include the prolonged use of powerful loop diuretics (see p. 757), osmotic diuresis (see Box 35-1) during poorly controlled diabetes mellitus, adrenal insufficiency with low aldosterone levels, and the recovery phase following acute renal failure or relief of urinary obstruction.
N40-1
Effect of Posture and Water Immersion on Na+ Excretion
Contributed by Gerhard Giebisch, Erich Windhager
On page 838, we introduce congestive heart failure as an example of the nonparallel behavior of ECF volume on the one hand and effective circulating volume on the other. Two additional examples that depend upon gravity are posture and water immersion.
Urinary Na+ excretion is lowest when one is standing (i.e., when thoracic perfusion is lowest), higher when one is lying down (recumbency), and highest when one is immersed up to the chin for several hours in warm water. During immersion, the hydrostatic pressure of the water compresses the tissues—and thus the vessels, particularly the veins—in the extremities and abdomen and consequently enhances venous return to the thorax. Recumbency—and, to a greater extent, water immersion—shifts blood into the thoracic vessels, increasing the so-called central blood volume (see p. 449). In contrast, the upright position depletes the intrathoracic blood volume. The thoracic vessels are immune to this compression because their extravascular pressure (i.e., intrapleural pressure; see p. 606) is unaffected by the water. Thus, it is the enhanced venous return alone that stimulates vascular sensors to increase Na+ excretion. This example clearly demonstrates that only special portions within the ECF compartment play critical roles in the sensing of ECF volume.
N40-2
Sensitivity of the Natriuretic Response to Increased Extracellular Fluid Volume
Contributed by Erich Windhager, Gerhard Giebisch
Figure 40-1B shows a hypothetical example of how urinary Na+ excretion (y-axis) changes in response to increases in isotonic extracellular water volume (upper x-axis) or amount of Na+ retained by the body (lower x-axis). In the example in the figure, the urinary Na+ excretion increases by 120 mmol/day for every 100 mmol of cumulative Na+ retention. This proportionality is indicated by the slope of the line. However, this slope need not be the same for every person.
In a patient with abnormal Na+ retention, the natriuretic response must be less sensitive than normal (i.e., the slope of the line in Fig. 40-1B must be less steep). In other words, in response to an increase in Na+ intake, the patient would have to accumulate more Na+ and water (i.e., he or she would have to become more volume expanded than would a normal person) in order to sufficiently stimulate the kidneys to elicit the natriuretic response necessary for coming into Na+ balance (i.e., achieving a steady state in which urinary excretion balances dietary intake).
Decreases in effective circulating volume trigger four parallel effector pathways to decrease renal Na+ excretion
Figure 40-2 shows the elements of the feedback loop that controls the effective circulating volume. As summarized in Table 40-2, sensors that monitor changes in effective circulating volume are baroreceptors located in both high-pressure (see pp. 534–536) and low-pressure (see pp. 546–547) areas of the circulation. Although most are located within the vascular tree of the thorax, additional baroreceptors are present in the kidney—particularly in the afferent arterioles (see p. 730)—as well as in the CNS and liver. Of the pressures at these sites, it is renal perfusion pressure that is most important for long-term regulation of Na+ excretion, and thus blood pressure, because increased resistance to renal blood flow (e.g., renal artery stenosis) causes sustained hypertension (Box 40-2). The sensors shown in Figure 40-2 generate four distinct hormonal or neural signals (pathways 1 to 4 in the figure).
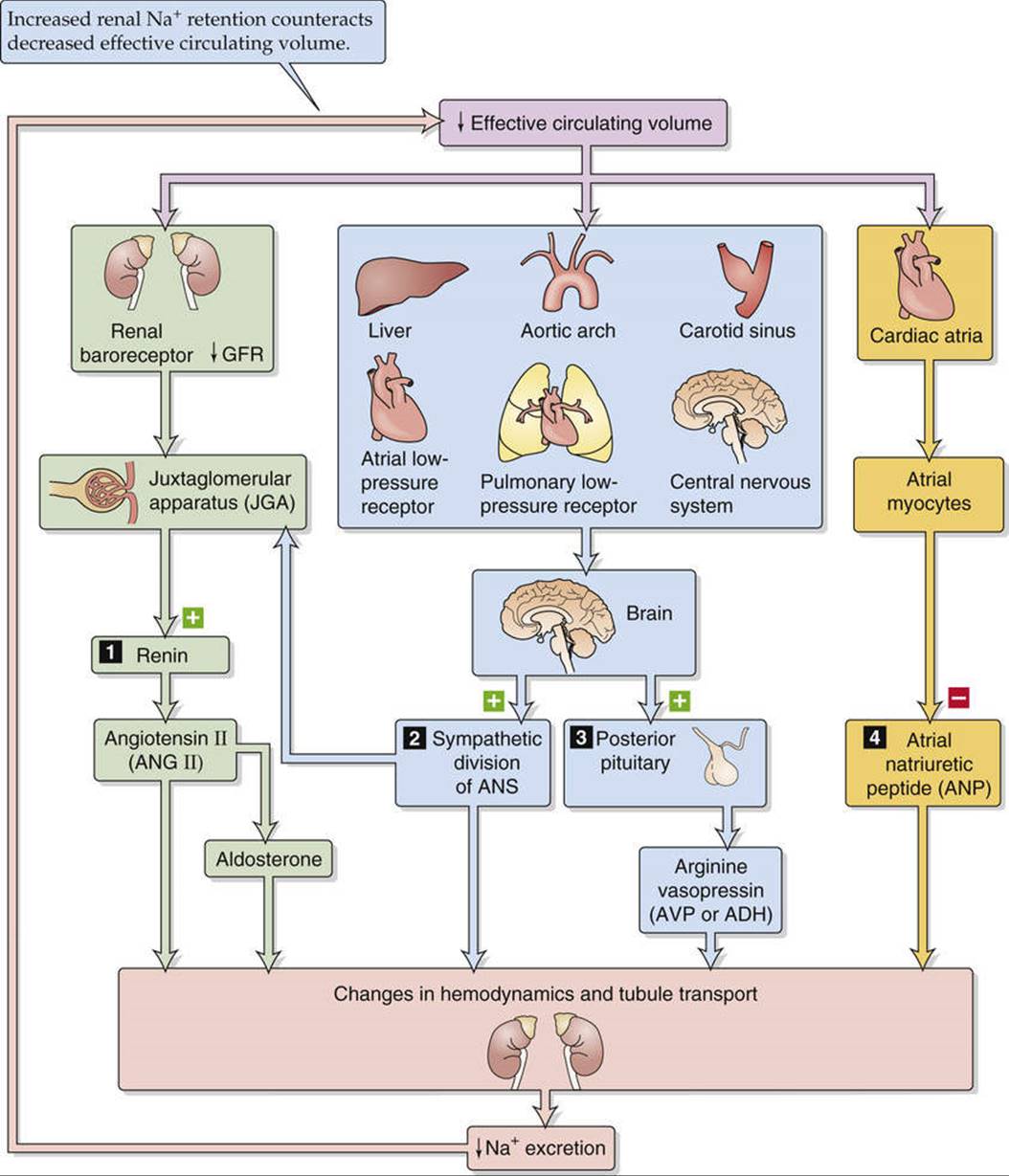
FIGURE 40-2 Feedback control of effective circulating volume. A low effective circulating volume triggers four parallel effector pathways (numbered 1 to 4) that act on the kidney, either by changing the hemodynamics or by changing Na+ transport by the renal-tubule cells. ANS, autonomic nervous system.
TABLE 40-2
ECF Volume Receptors
|
“Central” vascular sensors High pressure JGA (renal afferent arteriole) Carotid sinus Aortic arch Low pressure Cardiac atria Pulmonary vasculature Sensors in the CNS (less important) Sensors in the liver (less important) |
Box 40-2
Renal Hypertension
In the 1930s, Goldblatt produced hypertension experimentally in unilaterally nephrectomized animals by placing a surgical clip around the renal artery of the remaining kidney (one-kidney Goldblatt hypertension). The constriction can be adjusted so that it results not in renal ischemia, but only in a reduction of the perfusion pressure distal to the clip. This maneuver stimulates the renal baroreceptors, leading to a rapid increase in synthesis and secretion of renin from the clipped kidney. The renin release reaches a peak after 1 hour. As renin cleaves ANG I from angiotensinogen, systemic ANG I levels rise quickly. ACE, present mainly in the lungs but also in the kidneys, then rapidly converts ANG I into ANG II. Thus, within minutes of clamping the renal artery, one observes a sustained rise in systemic arterial pressure. The newly established stable elevation in systemic pressure then normalizes the pressure in the renal artery downstream from the constriction. From this time onward, circulating renin and ANG II levels decline toward normal over 5 to 7 days, while the systemic arterial pressure remains abnormally high. The early rise in blood pressure is the result of the renin-angiotensin vasoconstrictor mechanism, which is activated by the experimentally induced reduction in pressure and flow in the renal artery distal to the constriction. The later phase of systemic hypertension is the result of aldosterone release and of the retention of salt and water.
Unilateral partial clamping of a renal artery in an otherwise healthy animal also produces hypertension (two-kidney Goldblatt hypertension). As in the one-kidney model, the clipped kidney increases its synthesis and secretion of renin. Renin then causes ANG II levels to increase systemically and will, in addition to the effect on the clamped kidney, cause the nonclamped contralateral kidney to retain salt and water. As in the one-kidney model, the resulting hypertension has an early vasoconstrictive phase and a delayed volume-dependent phase. These models of hypertension show that the kidney can be critical as a long-term baroreceptor. Thus, when increased resistance in a renal artery leads to reduced intrarenal perfusion pressure, the rest of the body, including central baroreceptors, experiences—and cannot counteract—the sustained hypertension.
In both types of Goldblatt hypertension, administration of ACE inhibitors can lower arterial blood pressure. In fact, inhibiting ACE is therapeutically effective even after circulating renin and ANG II levels have normalized. The reason is that maintained hypertension involves an increased intrarenal conversion of ANG I to ANG II (via renal ACE), with the ANG II enhancing proximal Na+ reabsorption. Indeed, direct measurements show that, even after circulating renin and ANG II levels have returned to normal, the intrarenal levels of ACE and ANG II are elevated. ACE inhibitors lower systemic and intrarenal ANG II levels.
These experimental models correspond to some forms of human hypertension, including hypertension produced by renin-secreting tumors of the JGA and by all types of pathological impairment of renal arterial blood supply. Thus, coarctation of the aorta, in which the aorta is constricted above the renal arteries but below the arteries to the head and upper extremities, invariably leads to hypertension. Renal hypertension also results from stenosis of a renal artery, caused, for example, by arteriosclerotic thickening of the vessel wall.
In the first pathway, the kidney itself senses a reduced effective circulating volume and directly stimulates a hormonal effector pathway, the renin-angiotensin-aldosterone system, discussed in the section beginning on page 841. In addition, increased renal perfusion pressure itself can increase Na+ excretion independent of the renin-angiotensin-aldosterone system, as we shall see beginning on page 843.
The second and third effector pathways are neural. Baroreceptors detect decreases in effective circulating volume and communicate these via afferent neurons to the medulla of the brainstem. Emerging from the medulla are two types of efferent signals that ultimately act on the kidney. In one, increased activity of the sympathetic division of the autonomic nervous system reduces renal blood flow and directly stimulates Na+ reabsorption, thereby reducing Na+ excretion (discussed on pp. 842–843). In the other effector pathway, the posterior pituitary increases its secretion of AVP, which leads to conservation of water (discussed on p. 843). This AVP mechanism becomes active only after large declines in effective circulating volume.
The final pathway is hormonal. Reduced effective circulating volume decreases the release of atrial natriuretic peptide (ANP), thus reducing Na+ excretion (discussed on p. 843).
All four parallel effector pathways correct the primary change in effective circulating blood volume. An increase in effective circulating volume promotes Na+ excretion (thus reducing ECF volume), whereas a decrease in effective circulating volume inhibits Na+ excretion (thus raising ECF volume).
An important feature of renal Na+ excretion is the two-way redundancy of control mechanisms. First, efferent pathways may act in concert on a single effector within the kidney. For instance, both sympathetic input and hemodynamic/physical factors often act on proximal tubules. Second, one efferent pathway may act at different effector sites. For example, angiotensin II (ANG II) enhances Na+ retention directly by stimulating apical Na-H exchange in tubule cells (see Fig. 35-4) and indirectly by lowering renal plasma flow (see p. 746).
Increased activity of the renin-angiotensin-aldosterone axis is the first of four parallel pathways that correct a low effective circulating volume
The renin-angiotensin-aldosterone axis (Fig. 40-3) promotes Na+ retention via the actions of both ANG II and aldosterone. For a consideration of this axis in the context of the physiology of the adrenal cortex, see page 1029.
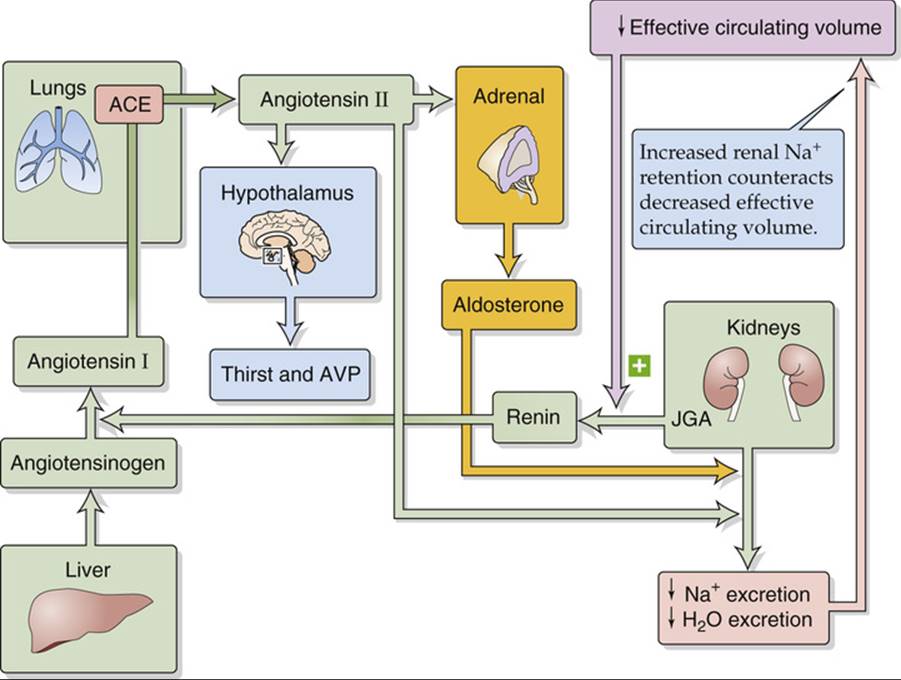
FIGURE 40-3 Renin-angiotensin-aldosterone axis.
Angiotensinogen, ![]() N23-12 also known as renin substrate, is an α2-globulin that is synthesized by the liver and released into the systemic circulation. The liver contains only small stores of angiotensinogen. Another protein, renin,
N23-12 also known as renin substrate, is an α2-globulin that is synthesized by the liver and released into the systemic circulation. The liver contains only small stores of angiotensinogen. Another protein, renin, ![]() N40-4 is produced and stored in distinctive granules by the granular cells of the renal juxtaglomerular apparatus (JGA; see p. 727). As discussed below (see p. 841), decreases in effective circulating volume stimulate these cells to release renin, which is a protease that cleaves a peptide bond near the C terminus of angiotensinogen, releasing the decapeptide angiotensin I (ANG I). Angiotensin-converting enzyme (ACE) rapidly removes the two C-terminal amino acids from the physiologically inactive ANG I to form the physiologically active octapeptide ANG II. ACE is present on the luminal surface of vascular endothelia throughout the body and is abundantly present in the endothelium-rich lungs. ACE in the kidney—particularly in the endothelial cells of the afferent and efferent arterioles, and also in the proximal tubule—can produce enough ANG II to exert local vascular effects. Thus, the kidney receives ANG II from three sources: (1) Systemic ANG II comes from the general circulation, originating largely from the pulmonary circulation. (2) Renal vessels generate ANG II from ANG I. (3) Proximal-tubule cells, which contain renin and ACE, secrete ANG II into its lumen. Both in the circulation and in the tubule lumen, aminopeptidases further cleave ANG II to the heptapeptides ANG III [ANG-(2-8)] and ANG-(1-7), which are biologically active.
N40-4 is produced and stored in distinctive granules by the granular cells of the renal juxtaglomerular apparatus (JGA; see p. 727). As discussed below (see p. 841), decreases in effective circulating volume stimulate these cells to release renin, which is a protease that cleaves a peptide bond near the C terminus of angiotensinogen, releasing the decapeptide angiotensin I (ANG I). Angiotensin-converting enzyme (ACE) rapidly removes the two C-terminal amino acids from the physiologically inactive ANG I to form the physiologically active octapeptide ANG II. ACE is present on the luminal surface of vascular endothelia throughout the body and is abundantly present in the endothelium-rich lungs. ACE in the kidney—particularly in the endothelial cells of the afferent and efferent arterioles, and also in the proximal tubule—can produce enough ANG II to exert local vascular effects. Thus, the kidney receives ANG II from three sources: (1) Systemic ANG II comes from the general circulation, originating largely from the pulmonary circulation. (2) Renal vessels generate ANG II from ANG I. (3) Proximal-tubule cells, which contain renin and ACE, secrete ANG II into its lumen. Both in the circulation and in the tubule lumen, aminopeptidases further cleave ANG II to the heptapeptides ANG III [ANG-(2-8)] and ANG-(1-7), which are biologically active.
N40-4
Renin Release from Granular Cells
Contributed by Erich Windhager, Gerhard Giebisch
As pointed out in the text, the granular cells are one of two cell types in which the exocytosis of a hormone decreases in response to a rise in [Ca2+]i. For example, if one raises [K+]o, the granular cell depolarizes. This depolarization probably opens voltage-gated Ca2+ channels (see p. 190) or decreases Ca2+ extrusion via an Na-Ca exchanger (see pp. 123–124). In either case, [Ca2+]i rises and blocks renin release. Similarly, applying Ca2+ ionophores—compounds that increase the permeability of the cell membrane to Ca2+—also raises [Ca2+]i and reduces renin release.
Increases in intracellular levels of cAMP have the opposite effect of raising [Ca2+]i—increases in [cAMP]i stimulate renin release from granular cells. Conversely, agents that inhibit adenylyl cyclase activity (e.g., β-adrenergic antagonists, α-adrenergic agonists, and A1 adenosine receptor agonists) decrease [cAMP]i and thereby inhibit renin release.
Reference
Kurtz A. Cellular control of renin secretion. Rev Physiol Biochem Pharmacol. 1989;113:1–38.
The principal factor controlling plasma ANG II levels is renin release from JGA granular cells. A decrease in effective circulating volume manifests itself to the JGA—and thus stimulates renin release—in three ways (see Fig. 40-2):
1. Decreased systemic blood pressure (sympathetic effect on JGA). A low effective circulating volume, sensed by baroreceptors located in the central arterial circulation (see p. 534), signals medullary control centers to increase sympathetic outflow to the JGA, which in turn increases renin release. Renal denervation or β-adrenergic blocking drugs (e.g., propranolol) inhibit renin release.
2. Decreased NaCl concentration at the macula densa (NaCl sensor). Decreased effective circulating volume tends to increase filtration fraction (the inverse of the sequence shown in Fig. 34-10), thereby increasing Na+ and fluid reabsorption by the proximal tubule (see p. 842) and reducing the flow of tubule fluid through the loop of Henle. Na+ reabsorption in the thick ascending limb (TAL) then decreases luminal [Na+] more than if tubular flow were higher. The resulting decrease in luminal [NaCl] at the macula densa stimulates renin release.
3. Decreased renal perfusion pressure (renal baroreceptor). Stretch receptors in the granular cells (see p. 727) of the afferent arterioles sense the decreased distention associated with low effective circulating volume. This decreased stretch lowers [Ca2+]i, which increases renin release and initiates a cascade that tends to promote Na+ reabsorption and thus increase blood pressure. Conversely, increased distention (high extracellular volume) inhibits renin release.
The above stimulation of renin release by a decrease in [Ca2+]i ![]() N40-4 stands in contrast to most Ca2+-activated secretory processes, in which an increase in [Ca2+]i stimulates secretion (see p. 221). Another exception is the chief cell of the parathyroid gland, in which an increase in [Ca2+]i inhibits secretion of parathyroid hormone (see pp. 1060–1061).
N40-4 stands in contrast to most Ca2+-activated secretory processes, in which an increase in [Ca2+]i stimulates secretion (see p. 221). Another exception is the chief cell of the parathyroid gland, in which an increase in [Ca2+]i inhibits secretion of parathyroid hormone (see pp. 1060–1061).
Intracellular cAMP also appears to be a second messenger for renin release. Agents that activate adenylyl cyclase ![]() N40-5 enhance renin secretion, presumably via protein kinase A. The question whether the effects of [cAMP]i and [Ca2+]i are independent or sequential remains open.
N40-5 enhance renin secretion, presumably via protein kinase A. The question whether the effects of [cAMP]i and [Ca2+]i are independent or sequential remains open. ![]() N40-3
N40-3
N40-3
Systemic versus Local Roles of the Juxtaglomerular Apparatus
Contributed by Emile Boulpaep, Walter Boron
The JGA performs two apparently opposite functions: maintaining a constant GFR (tubuloglomerular feedback, or TGF) and maintaining a constant whole-body blood pressure by modulating renin release. TGF (see pp. 750–751) is a local phenomenon, whereas the release of renin has systemic consequences (see pp. 841–842).
In the case of tubuloglomerular feedback (i.e., the local response), decreased renal perfusion pressure, reduced filtered load, or enhanced proximal fluid reabsorption all lead to a decrease in the flow of tubule fluid past the macula densa, as well as to a decrease in Na+ delivery and Na+ concentration. Within seconds after such a transient disturbance, and by an unknown mechanism, TGF dilates the afferent arteriole of the same nephron in an attempt to increase single-nephron glomerular filtration rate (SNGFR) and restore fluid and Na+ to that particular macula densa.
In the case of renin release (i.e., the systemic response), by contrast, a sustained fall in arterial pressure or a contraction of the extracellular volume reduces fluid delivery to many maculae densae, leading to the release of renin. Renin, in turn, causes an increase in local and systemic concentrations of ANG II. Besides causing general vasoconstriction, ANG II constricts the afferent and efferent glomerular arterioles, thereby decreasing GFR. This effect is opposite to that of TGF: TGF dilates a single afferent arteriole, whereas renin release constricts many afferent and efferent arterioles.
TGF may be viewed as a mechanism designed to maintain a constant SNGFR, whereas renin release is aimed at maintaining blood pressure by both systemic and renal vasoconstriction (i.e., hemodynamic effects), as well as by reducing SNGFR and enhancing tubule Na+ reabsorption (Na+-retaining effects). TGF is a minute-to-minute, fine control of SNGFR that can be superseded by the intermediate- to long-term effects of the powerful renin response, which comes into play whenever plasma volume and blood pressure are jeopardized. It must be emphasized that renin release is governed not only by the JGA but also by other mechanisms, in particular by changes in the activity of sympathetic nerves (see pp. 842–843).
N40-5
Other Factors that Activate Adenylyl Cyclase in Granular Cells
Contributed by Gerhard Giebisch, Erich Windhager
Agents that activate adenylyl cyclase in the granular cells of the JGA—and thus stimulate renin release—include forskolin, β-adrenergic agonists, A2 adenosine receptor agonists, dopamine, and glucagon. In addition, exogenous cAMP and phosphodiesterase inhibitors enhance renin secretion. All of these agents presumably act through protein kinase A.
Additional factors also modulate renin release. Prostaglandins E2 and I2 and endothelin all activate renin release. Agents that blunt renin release include ANG II (which represents a short feedback loop), AVP, thromboxane A2, high plasma levels of K+, and nitric oxide.
ANG II has several important actions as follows:
1. Stimulation of aldosterone release from glomerulosa cells in the adrenal cortex (see p. 1028). In turn, aldosterone promotes Na+ reabsorption in the distal tubule and collecting tubules and ducts (see p. 766).
2. Vasoconstriction of renal and other systemic vessels. ANG II increases Na+ reabsorption by altering renal hemodynamics, probably in two ways (Fig. 40-4). First, at high concentrations, ANG II constricts the efferent more than the afferent arterioles, thus increasing filtration fraction and reducing the hydrostatic pressure in the downstream peritubular capillaries. The increased filtration fraction also increases the protein concentration in the downstream blood and hence raises the colloid osmotic pressure of the peritubular capillaries. The changes in each of these two Starling forces favor the uptake of reabsorbate from peritubular interstitium into peritubular capillaries (see pp. 763–765) and hence enhance the reabsorption of Na+ and fluid by the proximal tubule. Second, ANG II decreases medullary blood flow through the vasa recta. Low blood flow decreases the medullary washout of NaCl and urea (see pp. 813–815), thus raising [urea] in the medullary interstitium and enhancing Na+ reabsorption along the thin ascending limb of Henle's loop (see p. 811).
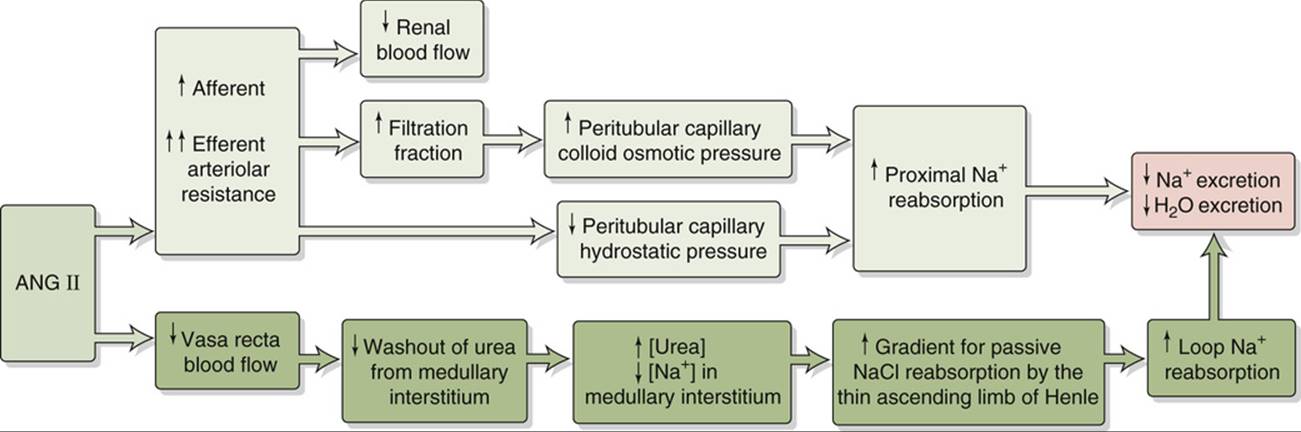
FIGURE 40-4 Hemodynamic actions of ANG II on Na+ reabsorption.
3. Enhanced tubuloglomerular feedback. ANG II raises the sensitivity and lowers the set-point of the tubuloglomerular feedback mechanism (see pp. 750–751), so that an increase in Na+ and fluid delivery to the macula densa elicits a more pronounced fall in the glomerular filtration rate (GFR).
4. Enhanced Na-H exchange. ANG II promotes Na+ reabsorption in the proximal tubule, TAL, and initial collecting tubule (see pp. 765–766).
5. Renal hypertrophy. Over a prolonged time, ANG II induces hypertrophy of renal-tubule cells.
6. Stimulated thirst and AVP release. ANG II acts on the hypothalamus, where it increases the sensation of thirst and stimulates secretion of AVP from the posterior pituitary, both of which increase total-body free water. This ANG II effect represents an intersection between the systems for regulating effective circulating volume and osmolality.
Increased sympathetic nerve activity, increased AVP, and decreased ANP are the other three parallel pathways that correct a low effective circulating volume
Renal Sympathetic Nerve Activity
The second of the four parallel effector pathways for the control of effective circulating volume is the sympathetic nervous system. Enhanced activity of the renal sympathetic nerves has two direct effects on Na+reabsorption (see pp. 766–768): (1) increased renal vascular resistance, and (2) increased Na+ reabsorption by tubule cells. In addition, increased sympathetic tone has an indirect effect—enhancing renin release from granular cells (see previous section). These multiple actions of sympathetic traffic to the kidney reduce GFR and enhance Na+ reabsorption, thereby increasing Na+ retention and increasing effective circulating volume.
In everyday life (i.e., the unstressed state), the role of sympathetic nerve activity in kidney function appears to be modest at best. However, sympathetic innervation may play a role during challenges to volume homeostasis. For example, low Na+ intake triggers reduced renal Na+ excretion; renal denervation blunts this response. Another example is hemorrhage, in which renal sympathetic nerves emerge as important participants in preserving ECF volume. Conversely, expansion of the intravascular volume increases renal Na+ excretion; renal denervation sharply reduces this response as well.
Arginine Vasopressin (Antidiuretic Hormone)
As discussed below (see p. 844), the posterior pituitary releases AVP primarily in response to increases in extracellular osmolality. Indeed, AVP mainly increases distal-nephron water permeability, promoting water retention (see pp. 817–818). However, the posterior pituitary also releases AVP in response to large reductions in effective circulating volume (e.g., hemorrhage), and secondary actions of AVP—vasoconstriction (see p. 553) and promotion of renal Na+ retention (see p. 768)—are appropriate for this stimulus.
Atrial Natriuretic Peptide
Of the four parallel effectors that correct a low effective circulating volume (see Fig. 40-2), ANP is the only one that does so by decreasing its activity. As its name implies, ANP promotes natriuresis (i.e., Na+excretion). Atrial myocytes synthesize and store ANP and release ANP in response to stretch (a low-pressure volume sensor; see p. 547). Thus, reduced effective circulating volume inhibits ANP release and reduces Na+ excretion. ANP plays a role in the diuretic response to the redistribution of ECF and plasma volume into the thorax that occurs during water immersion and space flight (see p. 1233).
Acting through a receptor guanylyl cyclase (see pp. 66–67), ANP has many synergistic effects (see p. 768) on renal hemodynamics and on transport by renal tubules that promote renal Na+ and water excretion. ![]() N40-6 Although ANP directly inhibits Na+ transport in the inner medullary collecting duct, its major actions are hemodynamic—increased GFR and increased cortical and medullary blood flow. ANP also decreases the release of renin, independently inhibits aldosterone secretion by the adrenal gland, and decreases release of AVP. In summary, a decrease in effective circulating volume leads to a fall in ANP release and a net decrease in Na+ and water excretion.
N40-6 Although ANP directly inhibits Na+ transport in the inner medullary collecting duct, its major actions are hemodynamic—increased GFR and increased cortical and medullary blood flow. ANP also decreases the release of renin, independently inhibits aldosterone secretion by the adrenal gland, and decreases release of AVP. In summary, a decrease in effective circulating volume leads to a fall in ANP release and a net decrease in Na+ and water excretion.
N40-6
Renal Sites of Action of Atrial Natriuretic Peptide
Contributed by Erich Windhager, Gerhard Giebisch
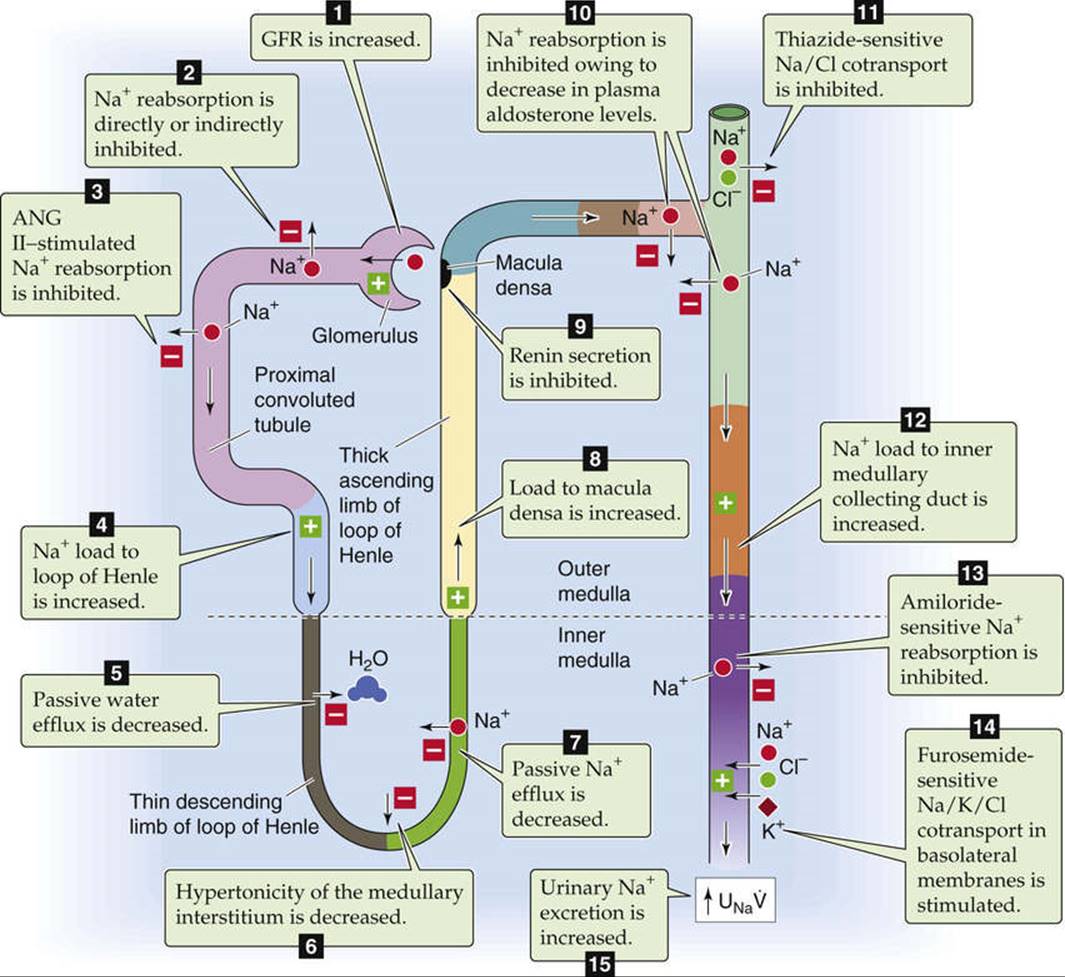
EFIGURE 40-1 Sites of action of ANP. UNa![]() , urinary sodium excretion rate. (Data from Atlas SA, Maack T: Atrial natriuretic factor. In Windhager E (ed): Handbook of Physiology, Section 8: Renal Physiology. New York, Oxford University Press [for American Physiological Society], 1992, pp 1577–1674.)
, urinary sodium excretion rate. (Data from Atlas SA, Maack T: Atrial natriuretic factor. In Windhager E (ed): Handbook of Physiology, Section 8: Renal Physiology. New York, Oxford University Press [for American Physiological Society], 1992, pp 1577–1674.)
High arterial pressure raises Na+ excretion by hemodynamic mechanisms, independent of changes in effective circulating volume
We have seen that expanding the effective circulating volume stimulates sensors that increase Na+ excretion via four parallel effector pathways (see Fig. 40-2). However, the kidney can also modulate Na+excretion in response to purely hemodynamic changes, as in the following two examples.
Large and Acute Decrease in Arterial Blood Pressure
If glomerulotubular (GT) balance (see p. 763) were perfect, decreasing the GFR would cause Na+ excretion to fall linearly (Fig. 40-5, blue line). However, acutely lowering GFR by partial clamping of the aorta causes a steep, nonlinear decrease in urinary Na+ excretion (see Fig. 40-5, red curve). When GFR falls sufficiently, the kidneys excrete only traces of Na+ in a small volume of urine. This response primarily reflects the transport of the classical distal tubule (see p. 765), which continues to reabsorb Na+ at a high rate despite the decreased Na+ delivery.
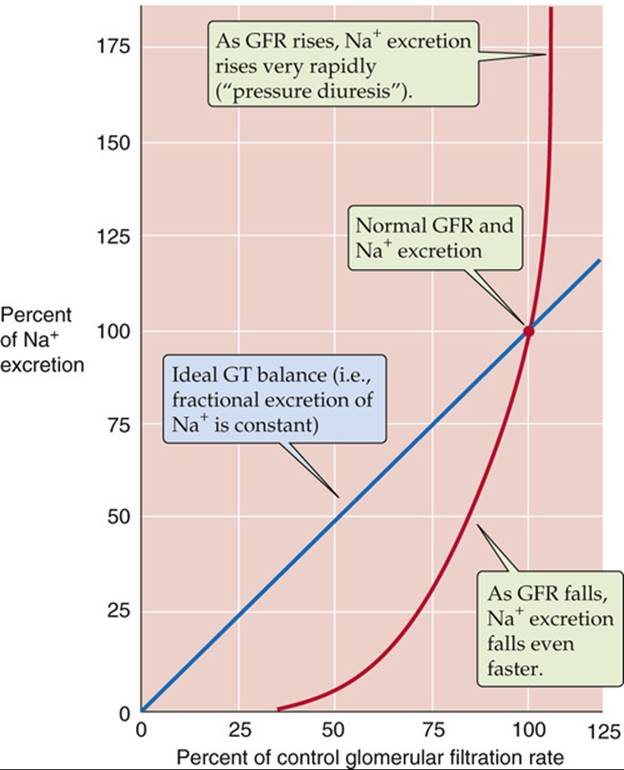
FIGURE 40-5 Effect of changes in GFR on urinary Na+ excretion. The blue line represents ideal glomerulotubular (GT) balance. The red curve summarizes data from dogs. The investigators reduced GFR by inflating a balloon in the aorta, above the level of the renal arteries. They increased GFR by compressing the carotid arteries and thus increased blood pressure. (Data from Thompson DD, Pitts RF: Effects of alterations of renal arterial pressure on sodium and water excretion. Am J Physiol 168:490–499, 1952.)
Large Increase in Arterial Pressure
In some cases, an increased effective circulating volume is accompanied by an increase in arterial pressure. Examples include primary hyperaldosteronism and Liddle disease ![]() N23-14, states of abnormally high distal Na+ reabsorption. The excess Na+ reabsorption leads to high blood pressure and compensatory pressure-induced natriuresis. One reason for this pressure diuresis is that hypertension increases GFR, increasing the filtered load of Na+, which by itself would increase urinary Na+ excretion (see Fig. 40-5, blue line). However, at least four other mechanisms contribute to the natriuresis (see Fig. 40-5, red curve). First, the increased effective circulating volume inhibits the renin-angiotensin-aldosterone axis and thus reduces Na+ reabsorption (see pp. 765–766). Second, the high blood pressure augments blood flow in the vasa recta, thereby washing out medullary solutes and reducing interstitial hypertonicity in the medulla (see pp. 813–815) and ultimately reducing passive Na+ reabsorption in the thin ascending limb (see p. 811). Third, an increase in arterial pressure leads, by an unknown mechanism, to prompt reduction in the number of apical Na-H exchangers in the proximal tubule. Normalizing the blood pressure rapidly reverses this effect. Finally, hypertension leads to increased pressure in the peritubular capillaries, thereby reducing proximal-tubule reabsorption (physical factors; see p. 763).
N23-14, states of abnormally high distal Na+ reabsorption. The excess Na+ reabsorption leads to high blood pressure and compensatory pressure-induced natriuresis. One reason for this pressure diuresis is that hypertension increases GFR, increasing the filtered load of Na+, which by itself would increase urinary Na+ excretion (see Fig. 40-5, blue line). However, at least four other mechanisms contribute to the natriuresis (see Fig. 40-5, red curve). First, the increased effective circulating volume inhibits the renin-angiotensin-aldosterone axis and thus reduces Na+ reabsorption (see pp. 765–766). Second, the high blood pressure augments blood flow in the vasa recta, thereby washing out medullary solutes and reducing interstitial hypertonicity in the medulla (see pp. 813–815) and ultimately reducing passive Na+ reabsorption in the thin ascending limb (see p. 811). Third, an increase in arterial pressure leads, by an unknown mechanism, to prompt reduction in the number of apical Na-H exchangers in the proximal tubule. Normalizing the blood pressure rapidly reverses this effect. Finally, hypertension leads to increased pressure in the peritubular capillaries, thereby reducing proximal-tubule reabsorption (physical factors; see p. 763).