Robert D. Winfield
Lyle L. Moldawer
Introduction
Gene therapy refers to the transfer or delivery of nucleic acids to somatic cells, resulting in a therapeutic effect through the correction of a genetic defect, the production of a therapeutically useful protein, or the attenuation of endogenous mRNA (1). While traditional medical and surgical treatment modalities are often effective in the treatment of disease processes, most small-molecule approaches are nonspecific, carrying with them the risks of adverse side effects and the possibility of harm to the patient (2). Even protein-based therapies, such as monoclonal antibodies or receptor antagonists, frequently have associated untoward side effects related to antigen recognition and activation of acquired immunity. Additionally, in disease processes such as inborn errors of metabolism, or chronic inflammatory or autoimmune diseases such as diabetes mellitus or rheumatoid arthritis, medication is frequently temporizing, and not curative, and must be taken indefinitely. In theory, gene therapy is a more tailored approach to disease, with results that are not only therapeutically successful, but also specific to a disease process, often lacking in significant side effects, and potentially curative in chronic disease states.
Unfortunately, while gene therapy offers these potential opportunities, in most cases, they remain only theoretical and unrealized, and have not yet been applied to practice. A number of significant practical hurdles exist to the use of gene therapy in the clinical arena, and, despite some 20 years of experimental work, few clinical successes have resulted. However, the prognosis for gene therapy is, in general, quite bright, and there have been a number of impressive results demonstrating the power and potential utility of this approach. This chapter will briefly review these research areas and summarize the essential aspects of human gene therapy, including the mechanism, possible risks, disease states amenable to gene therapy, and clinical use now and in the future. Our emphasis in the latter sections of this review will focus on the clinical application of gene therapy to acute inflammatory conditions in critical illness, such as sepsis, traumatic injury, and adult respiratory distress syndromes.
Overview
While the concept of gene therapy is a relatively simple one, the practice is complex and requires the following:
· The identification of a gene target in a disease of interest
· Creation of a DNA sequence that will generate a therapeutically active product
· Selection and incorporation of the sequence into an appropriately selected vector with an appropriate promoter sequence
· Delivery of the vector into the cells of interest
· The successful incorporation of the sequence into the host's cellular machinery for expression
A good deal of groundwork has been laid for the identification of gene targets and the creation of DNA sequences through efforts such as the recently completed Human Genome Project. Utilizing the information obtained through this and other undertakings, the number of areas for potential intervention has continued to multiply.
Vectors
With targets identified, the first step toward the integration of the gene sequence into the cells of interest is the selection of a vector. In this context, we use the term vector to refer to the delivery system for exogenous genetic material, and it can be as simple as a bacterial plasmid (Fig. 52.1) or as complicated as a recombinant virus. In general, we are not referring to the direct administration of nucleic acids with catalytic or other activities, such as ribozymes or small inhibitory RNA (siRNA) sequences. Rather, we are discussing expression systems where the host synthetic machinery is required to express either the induced protein or RNA sequences. Immediate goals for the creation and use of a vector are the delivery of genetic material to the correct site and expression of that material at a meaningful (therapeutic) level, all in a controlled fashion (3). No vector currently in use is completely successful at achieving this end. What follows is a discussion of a few of the more commonly considered vectors utilized at present, with a brief summary evaluation of some of their positive and negative features in critical illness.
Viral Vectors
Viral vectors represented the first vehicle utilized in gene therapy, and their use is still commonplace today. In general, viral vectors offer the advantages of intrinsic delivery methods combined with an inherent means for incorporating genetic material into the host cells (either episomal or with integration into the host genome). This takes advantage of the evolutionary success that viruses have attained in inserting their genetic material into eukaryotic cells and manipulating their expression machinery. Their overall disadvantages include generation of an immune response to varying degrees, as well as variable assimilation of genetic material into the host and its expression. In fact, the major challenge today with the use of viral vectors is controlling the host immune and other cellular processes that have evolved to control and ultimately eliminate viral infections.
|
|
||||||||||||
|
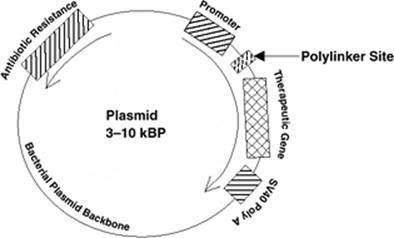
Figure 52.1. Prototypical expression plasmid used for nonviral gene therapy. Circular DNA generally based on a traditional bacterial plasmid (e.g., pBR), usually 3 to 10 kBP in size containing a promoter sequence (in this case, cytomegalovirus [CMV] early enhancer/promoter), the expressed transgene, an SV40 polyadenylation sequence, and one or more polylinker sites for splicing in and out DNA. In many cases, the plasmid may also contain a promoter and gene that conveys antibiotic resistance, which aids in the production of the plasmid in the bacterial host. |
||||||||||||
|
Table 52.1 Advantages and disadvantages of different gene therapy vectors |
||||||||||||
|
Viral vectors can also be divided based on whether they integrate their genetic information into the host genome (such as retroviruses and adeno-associated viruses (AAVs) or remain primarily episomal (such as adenovirus). While the former generally results in prolonged expression, often in excess of several months, the latter generally results in transient expression, lasting several days to a few weeks. While multiple different viruses have been proposed and are occasionally used as vectors, three primary viral vectors are in common use today, and will be presently discussed: adenoviruses, retroviruses, and AAVs.
Adenoviruses
Adenoviruses are the most commonly utilized vector in human gene therapy, accounting for 26% of all vectors currently being utilized in gene therapy clinical trials (4). Adenoviruses have a number of characteristics that account for their popularity in this field (Table 52.1):
1. They are easily grown in high viral titers.
2. They have a large capacity for transgene insertion.
3. They are efficiently transduced into both dividing and nondividing cells.
4. Only under very rare conditions do they incorporate into the host genome.
P.780
5. There are a variety of different serotypes with varying affinities for different cell types.
6. Many methods have been developed for manipulation of the adenoviral genome, allowing for tailored approaches to individual clinical scenarios and the potential for overcoming obstacles associated with immune responses and the duration of therapeutic activity (5).
The primary disadvantage of using adenovirus as a therapeutic vector is its intense activation of innate and both humoral and cellular immune responses. We and others have previously shown that administration of adenovirus into an immunocompetent host produces an often unwanted and dose-dependent induction of a number of proinflammatory cytokines, including tumor necrosis factor (TNF)-α (6,7). This activation of innate immunity and inflammation was most dramatically presented in the case of a subject who died from a “cytokine storm” and multisystem organ failure following the intravenous injection of adenovirus (8). In less dramatic but more frequent cases, adenovirus may activate inflammatory and immune processes, with the potential to prove harmful to the patient and undoubtedly limit expression of the therapeutic gene, and can prevent the effectiveness with repeated administration of the vector (9). Because of the ubiquitous nature of adenoviruses and because most patients have some existing acquired immunity to adenovirus infections, adenoviral vectors are often neutralized quickly in the setting of previous exposure. Furthermore, adenovirus receptors are present on many different types of cells, making targeting of specific cell types difficult, although this latter concern can be remedied to some degree with tissue-specific promoters (5,9). As a result, there are considerable ongoing research efforts to modify the adenoviral delivery system, in some cases by removing the adenoviral genes that, when expressed, are recognized by the host immune system. These “gutless” adenoviral recombinants reduce, but do not eliminate, their recognition by host immune tissues (10). Even with all of these limitations well known, the advantages of adenovirus still make it the most popular vector, in large part because of the high degree of, but transient, expression that is achieved. For the critically ill patient, adenovirus remains a potentially effective tool for the short-term delivery and expression of therapeutic proteins. As we will see, many of the hurdles associated with its inflammatory and immune processes can still be managed, even in the setting of acute inflammation.
Retroviruses
Retroviruses are second among the commonly utilized vectors (4) and are attractive for use in gene therapy, owing to the presence of three features:
· Membrane-coated viral particles are taken up through a receptor-mediated mechanism into target cells.
· A plus-stranded RNA genome is then incorporated into a double-stranded DNA within cellular chromosomes via reverse transcriptase.
· Particles are assembled in the cytoplasm with incorporation of the full-length retroviral mRNA as the mobile form of genetic information (11).
Clearly, these features could be beneficial, as the retrovirus offers a protective environment for delivery, an inherent mechanism for incorporation of genetic material into the host genome and the generation of both therapeutic product as well as mRNA for delivery to subsequent cells. As such, retroviruses can lead to a stable integration of genetic material into the host genome for long-lasting effects (1). Unfortunately, unlike the adenovirus, retroviruses cannot be generated in high titer. An additional complicating factor is that the cellular targets for retroviruses are quite limited, and retroviruses are unable to incorporate genetic material into cells that are not dividing. This is a significant limitation because adult mature cell populations are often not amenable to transduction. Finally, there is a small, but proven, risk of mutagenesis resulting from insertion of material into the host genome (12). This has been most dramatically demonstrated by the occurrence of leukemia in two patients with primary combined immunodeficiency treated with retrovirus-based gene therapy (13).
Adeno-associated Vectors
Adeno-associated viral vectors are less commonly employed in clinical trials than adenoviruses or retroviruses; however, their promise as a delivery method that can produce prolonged expression in the absence of a host inflammatory response has led to a recent increase in enthusiasm for their use. Adeno-associated virus is a replication-deficient parvovirus found in humans as well as nonhuman primates, and exists in over 100 distinct variants (14). The advantageous features of AAV are many, including its nonpathogenicity, long duration of infection, large number of variants, generally mild immunogenicity, ease of genomic modification, and the fact that a number of recombinant production and purification methods have been developed for it (15). A number of experimental studies have demonstrated long-term expression of a variety of genetic materials following transfection of recombinant AAV vectors into a number of tissues, including muscle, lung, liver, gut, the central nervous system, and eye (16). The primary limitation to the use of AAV is its small size, which limits the amount of material that may be packaged into the vector. A second potential drawback is the presence of a significant lag time between the administration of the AAV and maximal gene expression, which may limit its efficacy in conditions where a rapid response is desirable, as in the acute inflammatory conditions to be discussed later. Additionally, since the expression of genetic material is so durable when AAV is utilized, improved regulatory mechanisms are necessary to control expression in disease states where constitutive expression would be deleterious (16). Again, this is potentially problematic in acute inflammation, where long-term immune suppression or enhancement could be undesirable.
Nonviral Vectors
With the intrinsic problems associated with the use of viral vectors, it is not surprising that significant research has been directed toward the development of synthetic vectors that do not rely on viral delivery systems. Interestingly, the exploration of nonviral delivery systems goes back to the early 1970s when it was first shown that exogenous nucleic acids could be readily taken up into cells (17). Initially, it was believed in the mid-1990s that these nonviral vectors would have the potential to overcome the previously discussed issues of generated immune responses, nonspecificity, and potential mutagenesis associated with viral delivery systems. Additionally, the perfect nonviral vector would be incorporated efficiently into dividing and nondividing cells, and have a large DNA capacity (18). As with viral vectors, these ideals have not been fully realized. In fact, a number of significant hurdles remain that have limited the usefulness of current nonviral approaches. The primary difficulties with nonviral approaches are the recognition of plasmid DNA by components of the innate immune system and generation of an inflammatory response; at the same time, the incorporation of DNA tends to be poor, and their expression is limited. At present, the most commonly utilized nonviral vector is plasmid DNA, administered either as naked DNA or incorporated into liposomal delivery systems. An overview of these vectors follows.
Naked DNA
Naked DNA is appealing for use in human gene therapy because its introduction into a patient generally does not stimulate an acquired immune response. Unfortunately, plasmid DNA, generally composed of bacterial sequences, is often recognized by the innate immune system, and can generate a potentially limiting inflammatory response. Bacterial DNA generally has a methylation pattern distinct from eukaryotic DNA and, as such, is recognized by toll-like receptors, specifically TLR9. Activation of TLR9 by CpG sequences in plasmid DNA can induce a proinflammatory cytokine response via nuclear factor (NF)-κB–dependent signaling pathways (19). We have shown that the administration of plasmid DNA can exacerbate existing inflammation and increase mortality during acute inflammatory processes (20).
In addition, the administration of naked DNA is not target specific, nor does it have defenses against host nucleases. Under normal conditions, greater than 99% of administered DNA is destroyed by circulating, lysosomal, and cytosolic endonucleases (21); thus, the use of naked DNA generally relies upon the administration of relatively large amounts of DNA and novel approaches that can circumvent these limitations. A variety of methods have been introduced in an attempt to present naked DNA into host cells, as well as to prevent it from being destroyed. High-pressure delivery systems have been among the most studied and utilized (18). The process is a conceptually simple one, placing DNA onto a metallic microparticle and then using a “gene gun” to deliver the particles into target cells utilizing electromotive force (22). There is some evidence to suggest that uptake and incorporation of DNA is increased in injured or damaged cells, and these high-pressure delivery systems may inadvertently injure cell populations and induce cell recovery mechanisms. Unfortunately, though, while these mechanical delivery systems such as the gene gun deliver the DNA to cells of interest, they do so over a very limited area and have primarily been demonstrated to be effective only in superficial tissues; thus, different high-pressure methods, such as hydroporation (intravenous injection of a large volume of DNA in solution) and jet injection (intravenous injection of a small volume of DNA in solution), have been proposed to overcome these obstacles for the systemic delivery of DNA, and have each shown success in different animal and human models (23,24).
Electroporation is a technique that uses electrodes implanted in the tissue of interest to generate an electric field, with a resultant increase in permeability for the subsequently introduced DNA. While gene transfer is increased substantially through this technique, it is limited by the need for surgery to place electrodes and the tissue damage generated by the electric field (18); additionally, the exact mechanism for the success of electroporation has been called into question (25). More recently described techniques include laser beam gene transduction, ultrasonic gene delivery, magnetofaction (using magnetic nanoparticles to carry DNA to target tissues via direction by a magnetic field), and photochemical internalization (using light-sensitive endosomal vesicles to deliver DNA, and then lysing these vesicles with wavelength-appropriate light). Each of these methods has been shown to either increase the efficiency of gene transfer or increase expression in target tissues (reviewed in reference 18).
Liposomal Delivery
Liposomal delivery methods are the most common traditional approaches to nonviral DNA delivery, and have been utilized extensively since their introduction as one of the initial gene delivery methods (26). The concept is that the cationic lipid forms an electrostatic association with the DNA, leading to collapse of the anionic polymer, forming what is known as a lipoplex (18). These lipoplexes may be modified by the addition of ligands, antibodies, or other lipids in an attempt to improve target specificity, improve their stability, or decrease their toxicity. The lipoplexes fuse with the cellular membrane and are incorporated into endosomes. While liposomes do not trigger cellular immunity per se, they can activate the innate immune system, and their toxicity is the major limitation to their widespread use as a vector. Recent efforts have focused on diminishing the toxicity of these vectors, and two recent efforts have proven effective at lessening the immune response. The first, a simple staged procedure involving injection of liposome followed by injection of DNA—rather than injection of a formed lipoplex—led to a decrease in cytokine production as well as a reduced inflammatory response denoted by diminished neutropenia, lymphopenia, thrombocytopenia, and complement depletion with an increased transgene expression in the lung (27). The second method involves the creation of a “safeplex” containing anti-inflammatory entities such as glucocorticoids, nonsteroidal anti-inflammatory drugs (NSAIDs), or NF-κB inhibitors (28). The authors demonstrated significant suppression of inflammatory cytokine production via this method to go along with efficient gene delivery and expression.
Sepsis and Acute Inflammation
Inflammation is, in a general sense, a principal component of innate immunity and is the body's first reaction to an infectious or injurious agent (29). Under normal circumstances, this response is a well-orchestrated series of events designed to promote the isolation or destruction of a bacterial or viral invader, or healing from a traumatic insult. However, when unchecked, the activation of the innate immune system may trigger additional inflammatory responses in distant organ systems in what has been termed the systemic inflammatory response syndrome (SIRS) (29). When SIRS occurs in the setting of a microbial infection, it is defined as sepsis. Patients who develop SIRS may recover from this condition or progress to multiple organ dysfunction syndrome (MODS), in which they develop signs and symptoms of failure in multiple organ systems. These conditions take a heavy toll on affected patients, with significant morbidity and, in the case of sepsis, a mortality rate ranging from 25% to 80% (reviewed in reference 30).
Despite significant advances in both our understanding of the mechanisms underlying inflammation and in the resuscitation of critically ill patients with SIRS and sepsis, mortality rates from these conditions have not changed significantly over the past three decades. Current modalities utilized in the treatment of patients suffering from these disease states remain largely supportive or carry with them potentially dangerous side effects. Immune modulation through gene therapy carries a great deal of promise and would appear to be well suited for treatment of patients with these conditions; however, barriers still exist to the use of gene therapy clinically.
The Perfect Gene Therapy Vector
What would the perfect gene therapy vector look like for a critically ill patient, if technically possible? The vector would, when administered to a critically ill patient or organism, be rapidly taken up by the target cells and the transgene expressed quickly, within minutes to hours. Administration of the vector would not require any invasive procedure other than current critical care management, and the vector itself would be silent in terms of eliciting or exacerbating an inflammatory or immune response. Expression would be transient and could be turned off or silenced once the patient or animal was in a recovery mode. Finally, there would be no long-term consequences associated with the vector administration or expression of the transgene.
Clearly, the perfect vector does not presently exist, and one could argue that no single vector will be optimal for all critically ill patients. What is clearly obvious, however, is that all of the research to date has used vectors and delivery systems that were not optimal. Progress in gene therapy for the treatment of critically ill patients will remain dependent upon advances in the technology and science of gene therapy, as well as in defining the optimal therapeutic gene to be administered.
Over the past 15 years, significant efforts have been made to define the role of gene therapy in acute inflammatory conditions. In the following discussion, recent developments in gene therapy for acute endotoxemia and sepsis will be summarized.
Endotoxicosis
Most studies involving gene therapy for acute endotoxemia have focused on the immunomodulatory roles of interleukin-10 (IL-10). Our laboratory was the first to explore the possibilities of gene therapy in endotoxemia, demonstrating the beneficial effect of expressing either a soluble TNF receptor or the human IL-10 gene on mortality (31). In a subsequent study by Drazan et al., the authors also achieved inhibition of TNF-α and IL-1β by using an adenoviral vector to express viral IL-10 in the livers of neonatal mice; interestingly, though, they did not note significant modulation of IL-6 production (32). Xing et al. likewise used an adenoviral vector given intramuscularly to express murine IL-10, finding that they achieved expression not only at the site, but also in distant tissues; further, they showed a decrease in the levels of TNF-α as well as IL-6 (33). IL-10 gene transfer has also been shown to decrease endotoxin-induced pulmonary inflammation when given intratracheally (34). Finally, it has been demonstrated that pretreatment with an AAV expressing IL-10 also confers a survival advantage in mice undergoing an endotoxemic challenge, suggesting that it may be possible to pretreat susceptible individuals (35).
While IL-10 has received the most attention with gene therapy for the treatment of endotoxicosis, other targets have been explored. Among the first to utilize a different approach, Baumhofer et al. performed gene transfer of other anti-inflammatory cytokines, IL-4 and IL-13, using cationic liposomes, finding a decreased TNF-α response to a lethal endotoxin challenge and showing a survival advantage in experimental mice when compared with controls (36). Alexander et al. showed similar results utilizing an adenoviral vector expressing bacterial permeability increasing protein prior to lethal endotoxicosis (37).
Given the beneficial effects of IL-10–based gene therapy, there has been increasing interest in therapeutic approaches aimed at interfering directly with inflammation signaling pathways. One such approach has been to target nuclear factor-κB (NF-κB) activation during inflammation. For example, Matsuda et al. evaluated the effect of transfecting an NF-κB decoy into lung tissue and demonstrated decreased expression of inflammatory mediators in lung tissue, decreased pulmonary vascular permeability, and improved blood gas parameters when compared to control (38). A similar approach has been to overexpress another group of targeted proteins, the suppressor of cytokine signaling (SOCS) family, which has been determined to be a group of feedback inhibitors of cytokine receptor signaling. Fang et al. used a liposomal delivery system to transfer SOCS3 and IL-10 into mice that were subsequently given an endotoxic challenge. The authors reported that delivery of SOCS3 in addition to IL-10 led to the greatest improvement in survival, but that administration of either independently improved the survival of mice at 48 hours (39). Finally, Nakamura et al. found that they could diminish the degree of renal dysfunction typically seen in endotoxemia by using adenoviral-mediated delivery of β2-adrenoceptor to rat kidneys (40).
While these approaches have yielded generally positive results in animal models, concerns have been raised regarding the safety of administering a potentially immunogenic vector in the setting of acute inflammation. Our laboratory evaluated this possibility, showing that there is TNF-α–mediated hepatic injury when first-generation adenoviral vectors were administered in the setting of endotoxicosis (7); however, we also demonstrated that utilizing lower doses and incorporating an IL-10 construct into the vector abrogated this injury. These data were later supported by findings by Fejer et al., who demonstrated dramatic increases in TNF-α and nitric oxide levels in the kidney, liver, lung, and spleen. They further showed that these findings correlated with a diminished survival in animals treated with adenovirus and lipopolysaccharide (LPS) (41).
Sepsis
Sepsis is a particularly challenging disorder for treatment by gene therapy because sepsis is a systemic disease (29), and targeting therapy to a single tissue or organ is generally ineffective. In addition, the failure of most monotherapies for the treatment of sepsis has convincingly shown that the pathologic basis for sepsis is multifactorial, and treatment against a single component of sepsis is unlikely to be dramatically successful.
With that said, however, the primary effort of using gene therapy in sepsis has been directed against the exuberant inflammatory response; as in endotoxicosis, by far the most commonly used approach has been the forced expression of the anti-inflammatory protein, IL-10. IL-10 is a particularly attractive transgene in sepsis for a number of reasons (42,43). IL-10 is a profoundly anti-inflammatory cytokine and is known to suppress the expression of early proinflammatory genes like TNF-α and IL-1 by preventing NF-κB translocation. As previously discussed, IL-10 gene therapy has been frequently successful in lethal endotoxicosis. Importantly, IL-10 works through what is known as a “bystander effect,” meaning that transfection does not need to occur in every cell, since expression and secretion of IL-10 by a few transfected cells can produce biologic effects in adjacent, but not transfected, cells or tissues. Unfortunately, treatment of septic animals with the systemic administration of IL-10 protein has produced variable results, with most investigators unable to show any therapeutic benefit (44).
Because of the need for rapid but transient expression of the transgene, most transfection schemes have used either adenovirus or plasmid-based nonviral approaches for sepsis and acute inflammatory injury. Although it is difficult to compare results from different investigators with different vectors and delivery systems, the results have generally been positive with IL-10–based gene therapies in severe sepsis. Probably the most convincing data with systemic IL-10–based gene therapy has come from the recent studies of Kabay et al. in Turkey (45,46). These investigators used a plasmid-based approach with cationic liposomes, and demonstrated that the prior intraperitoneal injection of plasmids expressing human IL-10 could improve survival and reduce end-organ injury in a cecal ligation and puncture model of sepsis. The authors were able to demonstrate—using immunohistochemistry—human IL-10 expression in the liver, kidney, and lung following gene therapy.
We used plasmid-based IL-10 gene therapy for the treatment of experimental pancreatitis and were also able to show transient expression and improved outcomes (20,47,48). However, the data were complicated by the fact that the administration of cationic liposomes and the plasmid DNA without the transgene alone actually exacerbated the inflammatory response to pancreatitis and worsened outcome (20). In this case, expression of the IL-10 was required to dampen the inflammatory response not only to the experimental pancreatitis, but also to the administered plasmid and cationic liposomes.
During the initial studies with nonviral approaches and IL-10, we were disappointed by the low level of expression and its very transient nature (47,48). As a result, we switched early to the use of adenovirus to express the human IL-10 gene, with the idea that we could achieve therapeutic levels with the protein, with sufficiently low infection titers that would not exacerbate the already activated innate immunity. We were well aware of the studies of Doerschug et al., who had shown that administration of first-generation adenoviral recombinants during polymicrobial sepsis (cecal ligation and puncture) actually worsened survival (49). In fact, we demonstrated that the clearance of adenovirus from the lungs of mice was due to the TNF-mediated activation of innate and acquired immune processes (6). Importantly, expression of IL-10 reduced the inflammatory response to the adenovirus and prolonged expression (6,50).
In our initial studies, we administered first-generation adenoviral recombinants to mice intravenously prior to the induction of sepsis by either a cecal ligation and puncture or zymosan administration (51,52). In both cases, transfection occurred primarily in the liver and resulted in high circulating levels of human IL-10 (ng/mL). Although we did not see a worsening in outcome as was reported by Doerschug et al. (49), we did not see any improvements in outcome either, despite good expression of the transgene. The studies suggested to us that the systemic expression of IL-10 following the intravenous administration of the viral vector would be ineffective, similar to the systemic administration of the protein, as reported by Remick et al. (44).
We were surprised, however, when we saw that the compartmentalized expression of the IL-10 adenoviral vector was beneficial in both experimental models. Interestingly, the intratracheal administration of adenovirus expressing human or viral IL-10 improved outcome in the zymosan model of acute lung injury and multiorgan failure (51). Interestingly, better effects were seen with viral IL-10 expression, which appeared to be more tissue-associated than in the systemic circulation (50), and the beneficial effects of human IL-10 expression were strongly dose dependent (51,53). Actually, higher doses of adenovirus administered intratracheally and producing systemic concentrations of protein were frequently less effective than lower doses producing only local expression (53). Interestingly, the adenovirus also had to be administered prior to the inflammatory stimulus; once the process was ongoing, gene therapy was ineffective (51).
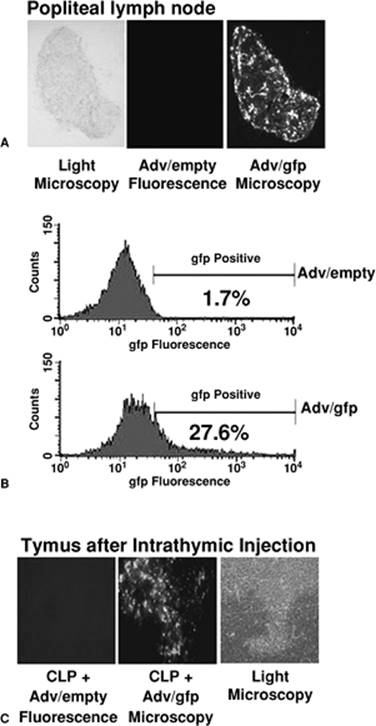
Also exciting was the observation that when very small quantities of adenovirus expressing human IL-10 were injected either subcutaneously into the footpad of mice or directly into the thymus, outcome from a cecal ligation and puncture-induced sepsis was markedly improved (52,54). Subsequent studies revealed that this compartmentalized administration of the adenoviral vector resulted in the transfection and expression of IL-10 by primarily dendritic cells (54,55). In the case of footpad injections, these IL-10 expressing dendritic cells then migrated to the draining lymph nodes where they expressed the protein in the context of class II expression and antigen presentation (Fig. 52.2). In these studies, we learned that autocrine production of IL-10 created a novel dendritic cell that had a phenotype consistent with an immature, tolerant, regulatory dendritic cell (56). Surprisingly, we subsequently showed that we could ex vivo transfect myeloid dendritic cells with this adenoviral recombinant expressing IL-10, and when they were reintroduced into a mouse prior to sepsis, could also improve outcome (56). Although the mechanism of protection is still unknown, it is speculated that this novel dendritic cell population expressing IL-10 may have fostered the expansion of regulatory T cells with the capacity of reducing the magnitude of the inflammatory response.
Although IL-10 has been the primary transgene used in models of sepsis as an anti-inflammatory agent, other approaches have been considered. As previously discussed, blocking NF-κB activation in endotoxicosis is an alternate approach aimed at preventing an exuberant inflammatory response (38); however, other approaches targeting more specific components of the injury response have been considered. For example, Weiss et al. used an adenoviral vector to delivery HSP70 into the lungs of mice following a cecal ligation and puncture (57). These investigators observed a significantly improved survival and reduced pulmonary inflammation.
|
|
|
Figure 52.2. Transfection efficiency following adenoviral gene transfer. A: An empty adenoviral vector or one expressing green fluorescent protein (gfp) was injected directly into the footpad of mice. Twenty-four hours later the popliteal lymph node was removed and green fluorescence determined by microscopy. Cells transfected with the adenovirus expressing gfp could be detected in accessory cells, predominantly dendritic cells. (Reprinted from Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83, with permission.) B: The same adenoviral vectors were injected into the thymus of mice, and 24 hours later, the thymi were removed, and gfp fluorescence determined in CD11c+ dendritic cells. Approximately 27% of the CD11c+ dendritic cells were expressing the gfp. C: Fluorescent microscopy confirms the gfp expression in the accessory cells of thymi injected with recombinant adenovirus expressing gfp. (Reprinted with permission from Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83.) |
Targeting the lung with adenovirus has been frequently used to deliver other therapeutic genes. Zhou et al. used adenovirus to deliver a mutant surfactant enzyme (CTP; phosphocholine cytidylyltransferase) to the lungs of mice prior to Pseudomonas infection and showed improved lung function and delayed mortality (58). Similar improvements in mortality and reduced lung injury were seen by Shu et al. when adenoviral expression of β-defensin-2 was also induced prior to Pseudomonas pneumonia (59). In both cases, the inflammatory potential of adenovirus did not appear to exacerbate the pneumonia response. Along these same lines, Chen et al. actually used an adenovirus to deliver TNF-α to the lungs of mice after a cecal ligation and puncture, but prior to a secondary Pseudomonas pneumonia (60). They hypothesized that the bacterial peritonitis might produce a defect in the pulmonary TNF response to the Pseudomonas, and adenoviral expression of TNF-α would improve host responses and outcome.
The Future of Gene Therapy for Critical Illness
Gene therapy for critical illness remains an elusive target in the future. There has been considerable progress made using gene therapy for the correction of inherited genetic disorders, and the possibility of cures for primary combined immunodeficiency, cystic fibrosis, α1-antitrypsin deficiency, Pompe disease, and other genetic diseases are on the horizon (Table 52.2). In contrast, progress as a drug (protein) delivery system for acute illnesses remains a considerable challenge. Limitations are primarily centered on optimizing the vector and promoter, which must still undergo further refinements to yield a delivery system that is rapid, transient, tissue specific, and safe, and one that does not exacerbate activated inflammatory and innate immune systems. Although adenovirus and plasmid-based delivery systems are most frequently used today, both have significant limitations in their current iterations.
In addition, there is little consensus about the optimal gene target to express, as critical illness, sepsis, trauma, and shock simultaneously affect a large number of immune and somatic cell systems, including innate and acquired immunity, thrombosis, fibrinolysis, acute phase, and neuron–endocrine, endocrine, and renal systems. Most approaches to date have been focused on either the global inflammatory response, the interaction between innate and acquired immunity, or specific lung functions. Whether a global approach will be superior to targeting specific components of the response in critical illness is still unproven.
Future Goals
Gene therapy remains an exciting and potentially important approach for the treatment of critically ill patients. It is unlikely that gene therapy will ever replace protein or small-molecule therapeutics, but because of its potential specificity and the ability to manipulate expression over extended periods of time, gene therapy remains an attractive but elusive goal for the treatment of the critically ill patient.
|
Table 52.2 Annotated summary of studies using gene therapy in models of critical illness |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
References
1. Rubanyi GM. The future of human gene therapy. Mol Aspects Med. 2001;22:113.
2. Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68:1781.
3. Anderson WF. Human gene therapy. Nature. 1998;392:25.
4. Vectors Used in Gene Therapy Clinical Trials. Vol. 2006.
5. McConnell MJ, Imperiale MJ. Biology of adenovirus and its use as a vector for gene therapy. Hum Gene Ther. 2004;15:1022.
6. Minter RM, Rectenwald JE, Fukuzuka K, et al. TNF-alpha receptor signaling and IL-10 gene therapy regulate the innate and humoral immune responses to recombinant adenovirus in the lung. J Immunol. 2000;164:443.
7. Oberholzer C, Oberholzer A, Tschoeke SK, et al. Influence of recombinant adenovirus on liver injury in endotoxicosis and its modulation by IL-10 expression. J Endotoxin Res. 2004;10:393.
8. Marshall E. Gene therapy death prompts review of adenovirus vector. Science. 1999;286:2244.
9. Ritter T, Lehmann M, Volk HD. Improvements in gene therapy: averting the immune response to adenoviral vectors. Biodrugs. 2002;16:3.
10. Kochanek S, Schiedner G, Volpers C. High-capacity ‘gutless’ adenoviral vectors. Curr Opin Mol Ther. 2001;3:454.
11. Baum C, Schambach A, Bohne J, et al. Retrovirus vectors: toward the plentivirus? Mol Ther. 2006;13:1050.
12. Nabel EG, Nabel GJ. Complex models for the study of gene function in cardiovascular biology. Annu Rev Physiol. 1994;56:741.
13. Marshall E. Gene therapy. Second child in French trial is found to have leukemia. Science. 2003;299:320.
14. Flotte TR. AAV-based gene therapy for inherited disorders. Pediatr Res. 2005;58:1143.
15. Monahan PE, Samulski RJ. AAV vectors: is clinical success on the horizon? Gene Ther. 2000;7:24.
16. Le Bec C, Douar AM. Gene therapy progress and prospects–vectorology: design and production of expression cassettes in AAV vectors. Gene Ther. 2006;13:805.
17. Bhargava PM, Shanmugam G. Uptake of nonviral nucleic acids by mammalian cells. Prog Nucleic Acid Res Mol Biol. 1971;11:103.
18. Conwell CC, Huang L. Recent advances in non-viral gene delivery. Adv Genet. 2005;53:3.
19. Zhao H, Hemmi H, Akira S, et al. Contribution of toll-like receptor 9 signaling to the acute inflammatory response to nonviral vectors. Mol Ther. 2004;9:241.
20. Norman J, Denham W, Denham D, et al. Liposome-mediated, nonviral gene transfer induces a systemic inflammatory response which can exacerbate pre-existing inflammation. Gene Ther. 2000;7:1425.
21. Wolff JA, Budker V. The mechanism of naked DNA uptake and expression. Adv Genet. 2005;54:3.
22. Yang NS, Burkholder J, Roberts B, et al. In vivo and in vitro gene transfer to mammalian somatic cells by particle bombardment. Proc Natl Acad Sci U S A. 1990;87:9568.
23. Zhang G, Gao X, Song YK, et al. Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther. 2004;11:675.
24. Walther W, Stein U, Fichtner I, et al. Low-volume jet injection for efficient nonviral in vivo gene transfer. Mol Biotechnol. 2004;28:121.
25. Liu F, Heston S, Shollenberger LM, et al. Mechanism of in vivo DNA transport into cells by electroporation: electrophoresis across the plasma membrane may not be involved. J Gene Med. 2006;8:353.
26. Felgner PL, Gadek TR, Holm M, et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A. 1987;84:7413.
27. Tan Y, Liu F, Li Z, et al. Sequential injection of cationic liposome and plasmid DNA effectively transfects the lung with minimal inflammatory toxicity. Mol Ther. 2001;3:673.
28. Liu F, Shollenberger LM, Huang L. Non-immunostimulatory nonviral vectors. Faseb J. 2004;18:1779.
29. Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83.
30. Efron P, Moldawer LL. Sepsis and the dendritic cell. Shock. 2003;20:386.
31. Rogy MA, Auffenberg T, Espat NJ, et al. Human tumor necrosis factor receptor (p55) and interleukin 10 gene transfer in the mouse reduces mortality to lethal endotoxemia and also attenuates local inflammatory responses. J Exp Med. 1995;181:2289.
32. Drazan KE, Wu L, Bullington D, et al. Viral IL-10 gene therapy inhibits TNF-alpha and IL-1 beta, not IL-6, in the newborn endotoxemic mouse. J Pediatr Surg. 1996;31:411.
33. Xing Z, Ohkawara Y, Jordana M, et al. Adenoviral vector-mediated interleukin-10 expression in vivo: intramuscular gene transfer inhibits cytokine responses in endotoxemia. Gene Ther. 1997;4:140.
34. Dokka S, Malanga CJ, Shi X, et al. Inhibition of endotoxin-induced lung inflammation by interleukin-10 gene transfer in mice. Am J Physiol Lung Cell Mol Physiol. 2000;279:L872.
35. Yamano S, Scott DE, Huang LY, et al. Protection from experimental endotoxemia by a recombinant AAV encoding interleukin 10. J Gene Med. 2001;3:450.
36. Baumhofer JM, Beinhauer BG, Wang JE, et al. Gene transfer with IL-4 and IL-13 improves survival in lethal endotoxemia in the mouse and ameliorates peritoneal macrophages immune competence. Eur J Immunol. 1998;28:610.
37. Alexander S, Bramson J, Foley R, et al. Protection from endotoxemia by adenoviral-mediated gene transfer of human bactericidal/permeability-increasing protein. Blood. 2004;103:93.
38. Matsuda N, Hattori Y, Takahashi Y, et al. Therapeutic effect of in vivo transfection of transcription factor decoy to NF-kappaB on septic lung in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1248.
39. Fang M, Dai H, Yu G, et al. Gene delivery of SOCS3 protects mice from lethal endotoxic shock. Cell Mol Immunol. 2005;2:373.
40. Nakamura A, Imaizumi A, Niimi R, et al. Adenoviral delivery of the beta2-adrenoceptor gene in sepsis: a subcutaneous approach in rat for kidney protection. Clin Sci (Lond). 2005;109:503.
41. Fejer G, Szalay K, Gyory I, et al. Adenovirus infection dramatically augments lipopolysaccharide-induced TNF production and sensitizes to lethal shock. J Immunol. 2005;175:1498.
42. Scumpia PO, Moldawer LL. Biology of interleukin-10 and its regulatory roles in sepsis syndromes. Crit Care Med. 2005;33:S468.
43. Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30:S58.
44. Remick DG, Garg SJ, Newcomb DE, et al. Exogenous interleukin-10 fails to decrease the mortality or morbidity of sepsis. Crit Care Med. 1998;26:895.
45. Kabay B, Kocaefe C, Baykal A, et al. 2006. Interleukin-10 gene transfer: prevention of multiple organ injury in a murine cecal ligation and puncture model of sepsis. World J Surg. 2007;31(1):200–209.
46. Kabay B, Kocaefe YC, Baykal A, et al. Liposome-mediated intraperitoneal interleukin 10 gene transfer increases survival in cecal litigation and puncture model of sepsis. Shock. 2006;26:37.
47. Denham W, Denham D, Yang J, et al. Transient human gene therapy: a novel cytokine regulatory strategy for experimental pancreatitis. Ann Surg. 1998;227:812.
48. Denham W, Yang J, MacKay S, et al. Cationic liposome-mediated gene transfer during acute pancreatitis: tissue specificity, duration, and effects of acute inflammation. J Gastrointest Surg. 1998;2:95.
49. Doerschug K, Sanlioglu S, Flaherty DM, et al. First-generation adenovirus vectors shorten survival time in a murine model of sepsis. J Immunol. 2002;169:6539.
50. Minter RM, Ferry MA, Rectenwald JE, et al. Extended lung expression and increased tissue localization of viral IL-10 with adenoviral gene therapy. Proc Natl Acad Sci U S A. 2001;98:277.
51. Minter RM, Ferry MA, Murday ME, et al. Adenoviral delivery of human and viral IL-10 in murine sepsis. J Immunol. 2001;167:1053.
52. Oberholzer C, Oberholzer A, Bahjat FR, et al. Targeted adenovirus-induced expression of IL-10 decreases thymic apoptosis and improves survival in murine sepsis. Proc Natl Acad Sci U S A. 2001;98:11503.
53. McAuliffe PF, Murday ME, Efron PA, et al. Dose-dependent improvements in outcome with adenoviral expression of interleukin-10 in a murine model of multisystem organ failure. Gene Ther. 2006;13:276.
54. Oberholzer A, Oberholzer C, Bahjat KS, et al. Increased survival in sepsis by in vivo adenovirus-induced expression of IL-10 in dendritic cells. J Immunol. 2002;168:3412.
55. Oberholzer C, Tschoeke SK, Bahjat K, et al. In vivo transduction of thymic dendritic cells with adenovirus and its potential use in acute inflammatory diseases. Scand J Immunol. 2005;61:309.
56. Oberholzer A, Oberholzer C, Efron PA, et al. Functional modification of dendritic cells with recombinant adenovirus encoding interleukin 10 for the treatment of sepsis. Shock. 2005;23:507.
57. Weiss YG, Maloyan A, Tazelaar J, et al. Adenoviral transfer of HSP-70 into pulmonary epithelium ameliorates experimental acute respiratory distress syndrome. J Clin Invest. 2002;110:801.
58. Zhou J, Wu Y, Henderson F, et al. Adenoviral gene transfer of a mutant surfactant enzyme ameliorates pseudomonas-induced lung injury. Gene Ther. 2006;13:974.
59. Shu Q, Shi Z, Zhao Z, et al. Protection against Pseudomonas aeruginosa pneumonia and sepsis-induced lung injury by overexpression of beta-defensin-2 in rats. Shock. 2006;26:365.
60. Chen GH, Reddy RC, Newstead MW, et al. Intrapulmonary TNF gene therapy reverses sepsis-induced suppression of lung antibacterial host defense. J Immunol. 2000;165:6496.