The Fundamentals of Recombinant DNA Technology: Molecular hybridization and DNA cloning
W. Stephen Kistler
Introduction
Our current ability to analyze and manipulate genomes began with reports in the 1970s of methods to cleave DNA at specific sites, to insert new DNA fragments into bacterial plasmids, and to sequence regions of DNA more than just a few nucleotides long. Prior to these developments genes in humans were known almost exclusively by their effects, that is from phenotypes and disease – genes were concepts, rather than structures. Gradually, it became possible to see exactly what a gene was and to to determine if genes were normal or mutated. A major step in the process was recognition that single strands of nucleic acids will form double stranded (ds) pairs with each other only if the sequences are highly similar or identical. Just as antibodies can detect single proteins in the midst of thousands of others, nucleic acids sequences will bind only to their complement in the presence of millions of non-matching sequences. A second major advance was the discovery of restriction enzymes, which convert chromosomal DNA into discrete fragments of useful length. Following separation by size, these smaller fragments could be detected by nucleic acid probes, and also sequenced. While many of the procedures used for analysis of DNA in the 20th century are mostly of historical interest today, some knowledge of them is necessary both for reading the older literature and for understanding modern recombinant DNA technology. The ability to splice and recombine fragments of DNA into viruses, bacterial plasmids, and even chromosomes has revolutionized the production of many clinically important human proteins as well as vaccines.
This Appendix provides an overview of some of the general techniques for forming so-called recombinant DNA and for cloning of this DNA.
The principles of molecular hybridization
Hybridization is based on the annealing properties of DNA
Hybridization is a process by which a piece of DNA or RNA of known nucleotide sequence, which can range in size from as little as 15 base pairs (bp) to several hundred kilobases, is used to identify a region or fragment of DNA containing complementary sequences. The first piece of DNA or RNA is called a probe. Probe DNA will form a complementary base pair with another strand of DNA, often termed the target, if the two strands are complementary and a sufficient number of hydrogen bonds are formed.
In molecular hybridization, it is essential that the probe and target are initially single-stranded
Probes can vary in both their size and their nature (DNA, RNA or oligonucleotide). However, one essential feature of any hybridization reaction is that both the probe and the target must be free to base pair with one another. For DNA hybridization, the two strands of DNA must first be separated by thermal or chemical treatment, a process called DNA denaturation ormelting. Once both probe and target DNA are single-stranded, mixing of the two under conditions that favor the formation of a double-stranded helix will allow complementary bases to recombine. This process is called DNA annealing or reassociation, and when a probe strand reacts with a target strand, the complex is termed a heteroduplex.
Formation of probe–target heteroduplexes is the key to the usefulness of molecular hybridization
The conditions under which DNA hybridization occurs and the reliability and specificity, or stringency, of hybridization are affected by several factors:
![]() base composition: GC pairs have three hydrogen bonds compared with the two in an AT pair. Double-stranded DNA with a high GC content is therefore more stable and has a higher meeting temperature (Tm).
base composition: GC pairs have three hydrogen bonds compared with the two in an AT pair. Double-stranded DNA with a high GC content is therefore more stable and has a higher meeting temperature (Tm).
![]() strand length: the longer a strand of DNA, the greater the number of hydrogen bonds between the two strands. Longer strands require higher temperatures or stronger alkali treatment to denature them; stability varies dramatically with length for very short probes but above a few hundred base pairs, stability is relatively insensitive to length and is determined primarily by base composition.
strand length: the longer a strand of DNA, the greater the number of hydrogen bonds between the two strands. Longer strands require higher temperatures or stronger alkali treatment to denature them; stability varies dramatically with length for very short probes but above a few hundred base pairs, stability is relatively insensitive to length and is determined primarily by base composition.
![]() reaction conditions: high cation concentration (typically Na+) favors double-stranded DNA (because the negative charges on the sugar-phosphate backbone are shielded from each other), while high concentrations of urea or formamide favor single-stranded DNA (because these reagents reduce base-stacking and can compete for hydrogen bond formation). Hybridizations are said to be carried out at low stringency when conditions strongly favor duplex formation, permitting some mismatch in the DNA duplex, and at high stringencywhen only matched, complementary duplexes are formed.
reaction conditions: high cation concentration (typically Na+) favors double-stranded DNA (because the negative charges on the sugar-phosphate backbone are shielded from each other), while high concentrations of urea or formamide favor single-stranded DNA (because these reagents reduce base-stacking and can compete for hydrogen bond formation). Hybridizations are said to be carried out at low stringency when conditions strongly favor duplex formation, permitting some mismatch in the DNA duplex, and at high stringencywhen only matched, complementary duplexes are formed.
Thus by appropriate selection of conditions (high stringency), a small 30–50 bp probe can require a perfect match to form a stable hybrid with its target. On the other hand, under low stringency a longer probe, e.g. 500 bp, might react with targets that contain multiple nucleotide mismatches or mutations (Fig. A2.1).
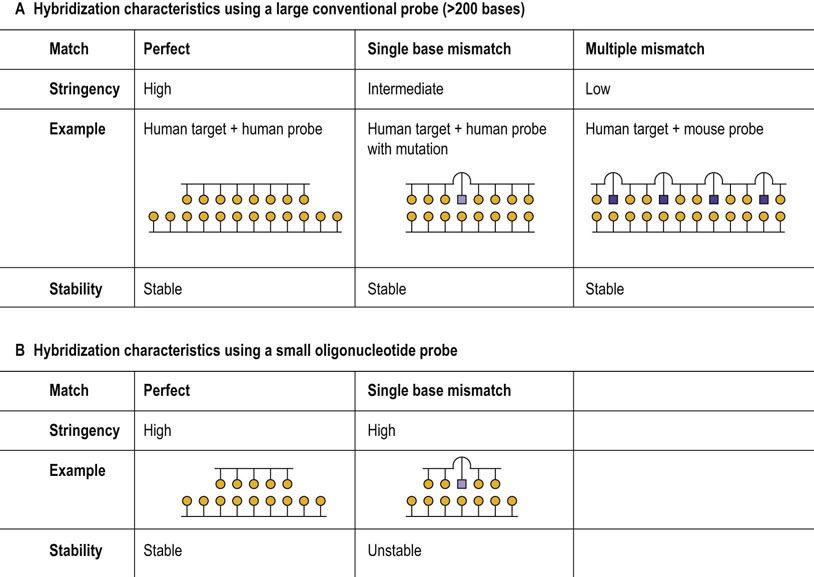
FIG. A2.1 Probe–template hybridization.
(A) Large probes, e.g. 200 bases or more, can form stable heteroduplexes with the target DNA even if there are a significant number of non-complementary bases in conditions of low stringency. (B) Oligonucleotide probes, in contrast, may discriminate between targets that differ by a single base under stringent conditions.
The stability of a nucleic acid duplex can be assessed by determining its melting temperature (Tm)
The melting temperature (Tm) is the temperature, in vitro, at which 50% of a double-stranded duplex has dissociated into single-strand form. For relatively long DNA probes, Tm is determined primarily by base composition, with AT-rich DNA melting at a lower temperature than GC-rich DNA. For humans and other mammals, the average GC content is about 40% and the melting temperature in moderate salt is about 87°C. For short oligonucleotides, such as the primers used in polymerase chain reactions (PCR), effects of length, composition and even the various dinucleotide sequences must be taken into consideration. This is because double-stranded DNA is stabilized by the degree of overlap by the stacked bases in successive nucleotides, and this varies depending on the specific nucleotide neighbors of a particular base. Computer programs are widely available to predict Tm values.
Probes must have a label to be identified
Implicit in the use of probes to identify pieces of complementary DNA is the notion that if hybridization occurs, the heteroduplex can be specifically detected. Thus, the probe is labeled so that the probe–target duplex can be identified. The labels generally fall into two categories, either isotopic, i.e. involving radioactive atoms, or nonisotopic, e.g. end-labeling probes with fluorescent tags or small ligand molecules. Many techniques involving probe hybridization and labeling still use radioisotopes such as 32P, 35S or 3H and, as such, require a method for detecting and localizing the radioactivity. The most common method involves the process of autoradiography. Autoradiography allows information from a solid phase, e.g. a gel or fixed-tissue sample, to be detected and saved in two-dimensional form as an exposed photographic image.
Southern blots are the prototype for methods that use specific hybridization probes to identify sequences in DNA or RNA
One of the fundamental steps in the evolution of molecular biology was the discovery that DNA could be transferred from a semisolid gel onto a nitrocellulose membrane in such a way that the membrane could act as a record of the DNA information in the gel and could be used for multiple-probe experiments. The process whereby the DNA is transferred to the membrane was first described by Edward Southern, but subsequent techniques based on the transfer of RNA and proteins have also adopted the direction theme and are called Northern (RNA target) and Western (protein target) blots, respectively.
Restriction Enzymes
Use of restriction enzymes to analyze genomic DNA
Restriction enzymes cleave DNA at specific nucleotide sequences
Restriction endonucleases cleave double-stranded DNA. These enzymes are sequence specific and each enzyme acts at a limited number of sites in DNA called ‘recognition’ or ‘cutting’ sites. Restriction endonucleases are part of the bacterial ‘immune system’. Bacteria methylate their own DNA, protecting it from their own restriction enzymes, but cleave unmethylated infecting viral or bacteriophage DNAs at specific sites, thereby inactivating the virus and restricting viral infection.
If DNA is digested by a restriction enzyme, the DNA will be reduced to fragments of varying sizes depending on how many cutting sites for that restriction enzyme are present in the DNA. The cutting sites are frequently palindromic sequences, sites at which the base sequence reads the same, backward and forward. It is important to note that each enzyme will cut DNA into a unique set of fragments (Fig. A2.2). Many restriction enzymes recognize sites that are typically four (e.g. HaeIII), six (e.g. EcoR I) or eight nucleotides (e.g. NotI) in length (Table A2.1). Variation in just one nucleotide within the recognition sequence makes a sequence completely resistant to a particular enzyme.
Table A2.1
Restriction endonucleases in common use
|
Endonuclease |
Restriction site |
Ends |
|
HaeIII |
GG*CC |
Flush |
|
CC*GG |
||
|
MspI |
C*CGG |
Sticky |
|
GGC*C |
||
|
EcoRV |
GAT*ATC |
Flush |
|
CTA*TAG |
||
|
EcoRI |
G*AATTC |
Sticky |
|
CTTAA*G |
||
|
NotI |
GC*GGCCGC |
Sticky |
|
CG CCGG*CG |
Enzymes can cleave DNA to produce ‘flush ends’ where the DNA is cut ‘vertically’ leaving two ends that do not have any overhanging nucleotides. However, if the DNA is cleaved ‘obliquely’, the DNA will have short single-stranded overhangs. Such ends are called ‘sticky’ because they will selectively rejoin (hybridize) to matching overhangs. The sites of cleavage of DNA by restriction enzymes are often described as palindromic because of their inverted repeat symmetry – they have identical sequences in opposite directions on the complementary strands.
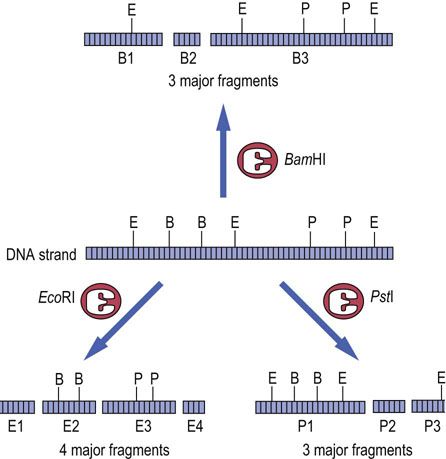
FIG. A2.2 Restriction enzyme digestion of DNA.
Digestion of a DNA molecule by several different restriction enzymes may result in many different fragments, even though the apparent size of the fragments is similar. For example, fragments E1 and P3 are of similar size but are clearly different pieces of DNA. E, EcoRI site; B, BamHI site; P, PstI site.
The frequency of the cutting sites for various enzymes varies with the length of the recognition site. Cut sites for an enzyme with a 4-base recognition site, such as HaeIII, would occur by chance once per 256 base pair sequence. Cut sites for an enzyme with an 8-base recognition sites, such as NotI, wold occur only once in about 656,000 base pairs. Thus, frequent cutters typically generate many small fragments, while rare cutters generate fewer and larger fragments. These differences can be exploited in the analysis of gene structure and chromosomal location.
DNA fragments, blotted onto a solid gel phase, are used as a template for exposure to a range of molecular probes
If DNA is digested by a restriction enzyme, the resulting digest can be separated on the basis of size by gel electrophoresis. Agarose gel electrophoresis is commonly used to separate fragments ranging in size from 100 bases to approximately 20 kb in length (above 40 kb, resolution is minimal). Following electrophoresis, the gels are soaked in a strong alkali solution to denature the DNA. These single-stranded fragments can then be transferred to a nitrocellulose or nylon membrane to which they bind readily and, if preserved properly, permanently. The process of transfer involves the passage of solute through the gel, passively carrying the DNA and producing an image of the gel on the membrane (Fig. A2.3). The membrane may then be probed with an oligonucleotide or DNA fragment (Southern blot), e.g. for genotyping, paternity testing or identification of cells incorporating a gene during a cloning experiment (below).
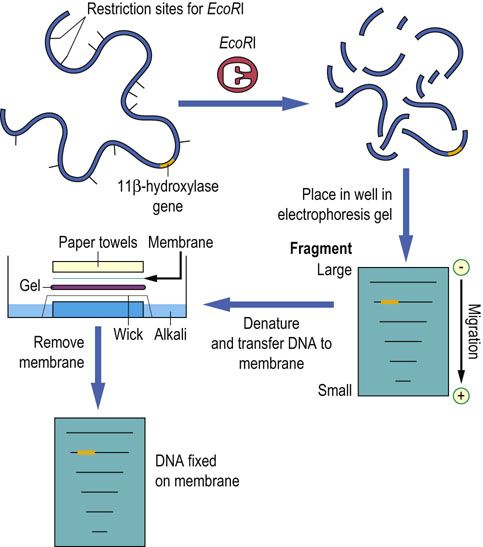
FIG. A2.3 Southern blotting of DNA.
DNA digested with a restriction enzyme is size-fractionated by agarose electrophoresis. The agarose gel is then placed in alkali to denature the DNA. The now single-stranded DNA can pass from the gel to the membrane (typically nylon or nitrocellulose) as buffer solution flows upward by capillary action, forming a permanent record of the digested DNA.
Restriction fragment length polymorphisms (RFLPs)
Analysis of restriction fragment length may be used to detect a mutation or polymorphism in a gene
If a change in DNA sequence creates or destroys a recognition site that yields a fragment detected by a probe, then the altered length of that fragment can be detected by Southern blotting. If a cleavage site is created, the fragment becomes smaller; if the cleavage site is eliminated, the fragment becomes larger. The different patterns generated as a result of a mutation or gene variant are known as restriction fragment length polymorphisms (RFLPs) (Fig. A2.4). Such RFLPs can be used either to identify disease-causing mutations, because of a single point mutation creating or abolishing a restriction site, or to study variation in noncoding DNA for use in the study of genetic linkage.
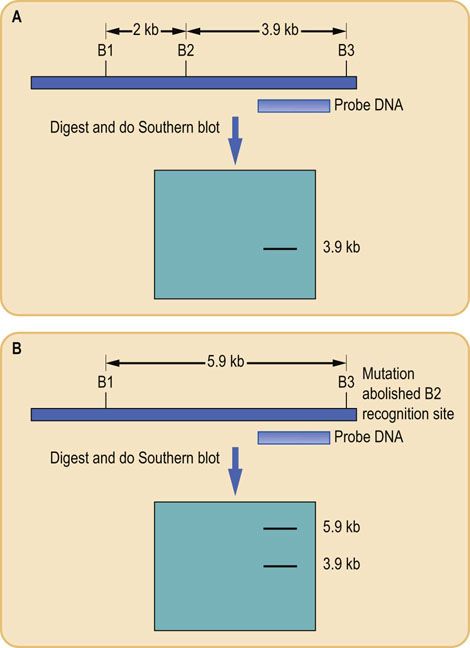
FIG. A2.4 Restriction fragment length polymorphisms (RFLP).
Variations in the nucleotide sequence of DNA, either due to natural variation in individuals or as a result of a DNA mutation, can abolish the recognition sites for restriction enzymes. This means that when DNA is digested with the enzyme whose site is abolished, the size of the resulting fragments is altered. Southern blotting and probe hybridization can be used to detect this change. Results are shown for a representative gene from (A) homozygous normal and(B) heterozygous mutant individuals. B, BamHl restriction site.
RFLP analysis can also detect larger pathologic changes in the DNA sequence, either deletions or duplications. Large deletions of a gene may abolish restriction sites; this leads to the disappearance of a fragment on a Southern blot in homozygous individuals. Alternatively, if a DNA duplication event occurs, a new gene may be formed, which has a different pattern of restriction sites that allow detection of the new gene. This type of hybridization is performed using large probes (0.5–5.0 kb) and is performed under moderate stringency, i.e. it is sufficiently rigid to allow hybridization of probe and target but also to tolerate minor differences, e.g. in noncoding DNA.
Low stringency hybridization of a probe to a Southern blot of digested DNA may allow genes related to, but not identical to, the starting gene to be identified. Many genes exist in families, or have nonfunctional, nearly identical copies elsewhere in the genome (pseudogenes), and thus hybridization of a probe may identify one or more restriction fragments, corresponding to related genes. Similarly, related genes in different species may be identified by using a single probe that can hybridize at low stringency to complementary sequences in blots of DNA from mouse, rat or other species.
![]() Clinical box Use of RFLPs for detection of the sickle cell gene
Clinical box Use of RFLPs for detection of the sickle cell gene
A 24-year-old Afro-Caribbean woman was referred for prenatal counseling. Her younger brother had sickle-cell anemia, and she had become pregnant. Her partner was known to be a carrier of the sickle-cell mutation (sickle-cell trait) and she wanted to know if her child would develop sickle-cell anemia.
Since the patient is at risk of being a carrier, she opted to have chorionic villus sampling (CVS) performed to detect the presence or absence of the sickle-cell mutation in her child. Analysis of her own DNA revealed that she was a carrier, and the CVS showed that the child was also a carrier and would not develop sickle-cell anemia.
Comment.
Occasionally, a mutation will directly abolish or create a restriction site and thus allow the use of a restriction-based method to demonstrate the presence or absence of the mutant allele. One widely examined mutation is the A > T substitution at codon 6 in the sequence for the β-globin gene responsible for sickle-cell disease (see Chapter 5). This results in a glutamine-valine (Glu-Val) mutation in the amino acid sequence β-globin and also abolishes a recognition site for MstII (CCTN(A > T)GG) in the β-globin gene. Digestion of normal human DNA with MstII and probing the Southern blot with a probe specific for the promoter of the β-globin gene yields a single band of 1.2 kb, as the nearest MstII site is 1.2 kb upstream in the 5'region of the gene. The abolition of the codon 6 restriction site means that the fragment size seen when probing MstII digested DNA is now 1.4 kb, as the next MstII site is located 200 bases downstream in the intron after exon 1. Thus, patients with sickle-cell anemia will show only one band, 1.4 kb, while carriers will have 2 bands, one 1.4 kb and another, 1.2 kb, and unaffected individuals will have a single 1.2 kb band.
Cloning of DNA
Cell-based cloning
Bacterial plasmids are bio-engineered to optimize their use as vectors
Cell-based cloning relies on the ability of replicating cells, e.g. bacteria, to permit replication of so-called recombinant DNA within them. Recombinant DNA refers to any DNA molecule that is artificially constructed from two pieces of DNA not normally found together. One piece of DNA will be the target DNA that is to be amplified and the other will be the replicatingvector or replicon, a molecule capable of initiating DNA replication in a suitable host cell.
Today, the majority of cell-based cloning is performed using bacterial cells. In addition to the bacterial chromosome, bacteria may contain extrachromosomal double-stranded DNA that can undergo replication. One such example is the bacterial plasmid. Plasmids are circular, double-stranded DNA molecules that undergo intracellular replication and are passed vertically from the parent cell to each daughter cell. However, unlike the bacterial chromosome, plasmids used in these techniques are copied many times during each cell division. Thus, plasmids are ideal carriers for the amplification of target DNA, and thus the encoded protein. Methods involving the use of plasmids are widespread throughout molecular biology.
Target DNA is introduced into a plasmid by using restriction enzymes to cut target and plasmid DNA so that the target DNA and the linearized vector DNA will have complementary sticky ends (Fig. A2.5). DNA ligase then covalently joins the target to the ends of the vector to form a closed circular recombinant plasmid. Once the target DNA is incorporated into the plasmid vector, the next step is to introduce the plasmid into a host cell to allow replication to occur. The cell membrane of bacteria is selectively permeable and prevents the free passage of large molecules such as DNA in and out of the cell. However, the permeability of cells can be altered temporarily by factors such as electric currents (electroporation) or high-solute concentration (osmotic stress), so that the membrane becomes temporarily permeable and DNA can enter the cell. Such a process renders the cells competent, i.e. they can take up foreign DNA from the extracellular fluid, a process known as transformation. This process is generally inefficient, so that only a small fraction of cells may take up plasmid DNA, and often only a single plasmid per bacterium is introduced during transformation. However, it is this process of cellular uptake of plasmid DNA that forms a critical step in cell-based cloning. Individual recombinant DNAs are easily resolved from one another because they are taken up by separate cells that can be isolated simply by spreading them on an agar surface.
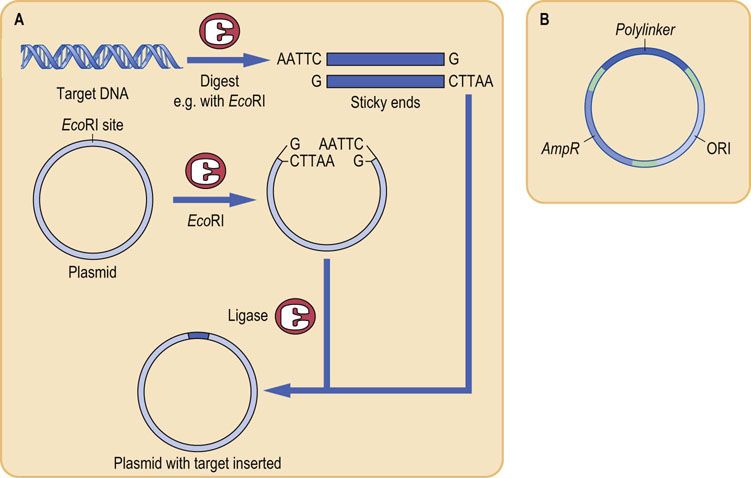
FIG. A2.5 Formation of a plasmid containing a target gene for cloning.
(A) DNA containing the target gene is digested with a restriction enzyme that will produce ‘sticky ends’, e.g. EcoRI. The plasmid also has a restriction site for EcoRI, so that when digested with EcoRI it becomes a linear DNA strand with ‘sticky ends’ complementary to the target. Upon ligation, the target and vector form a recombinant molecule. (B) Structure of a typical plasmid. The plasmid contains a gene conferring resistance to the antibiotic ampicillin (AmpR), and a polylinker region containing approximately 10 restriction enzyme recognition sites, which serve as sites for insertion of target DNA. The plasmid also contains ORI, the site for origin of DNA replication.
Following transformation, the cells are allowed to replicate, usually on a standard agar plate containing a suitable antibiotic (see Fig. A2.5B) to kill cells that do not harbor the plasmid containing the antibiotic resistance gene. This selection or screening process, based on antibiotic resistance, is an important step because of the low efficiency of the uptake of plasmid DNA into bacteria. Colonies (clones of single cells) are then ‘picked’ and transferred to tubes for growth in liquid culture and a second phase of exponential increase in cell number. This work is done automatically in microplate systems, so that, from a single cell and a single molecule of DNA, an extremely large number of cells containing multiple, identical recombinant plasmids can be generated in a relatively short time (Fig. A2.6). Recovery of the plasmid DNA is easy because it is a small, covalently intact circle, readily separated from the bacterial chromosomal DNA by a variety of techniques.
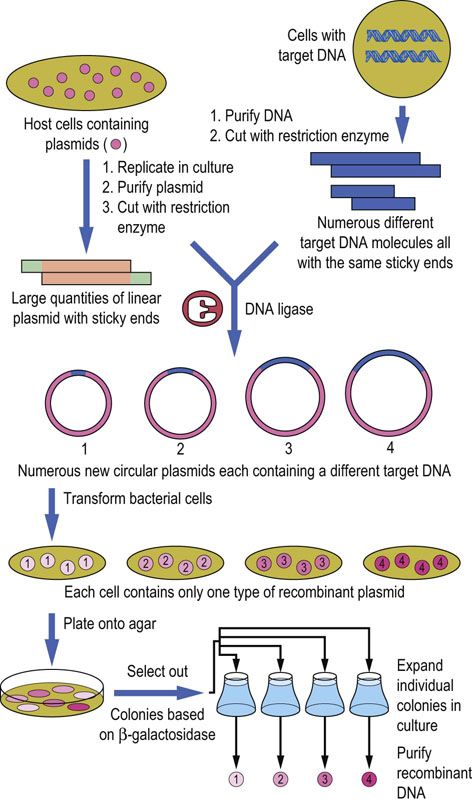
FIG. A2.6 Cell-based DNA cloning.
An example of cloning genomic DNA using bacterial cells. In general, each transformed bacterium will take up only a single plasmid molecule. Therefore individual bacterial colonies will contain many identical copies of just one particular recombinant DNA.
The bacterial cell is then grown in culture and the target protein is recovered following lysis of the cells. Alternatively, the plasmid might encode a protein with a signal sequence so that it is secreted into the medium; the signal sequence would be removed afterwards.
The technology for producing protein pharmaceuticals is a complex, multi-step process, often protected by trade secrets. For the bacterial synthesis of recombinant insulin (see Box onp. 615), for example, there is no gene or mRNA for the sequence of insulin – insulin is synthesized as preproinsulin, which is processed in pancreatic β-cells to yield the secreted hormone (see Chap. 21). The synthesis of human insulin in a bacterial system might involve the incorporation of the proinsulin gene into a plasmid, transformation into a bacterial host, synthesis of proinsulin, spontaneous disulfide crosslinking, processing by endopeptidases to remove the C-peptide, then folding to produce the active insulin molecule. The proteolytic processing might happen in the cell or following isolation of the proinsulin; intracellular processing would require encoding the protease in the bacterial plasmid. Other strategies might be imagined, such as synthesis of the A and B chains in separate bacterial hosts, then extracellular association into the active hormone. It might also be possible to design a protein product that would be secreted from the bacterial cell, then processed ex vivo to remove the secretory sequence and C-peptide.
Future Directions
Cloning of DNA is a rapidly evolving field in biomedical research and modern medicine. It is the basic methodology for production of genetically modified organisms (GMO), including agricultural products and transgenic and knock-out animals. More sophisticated eukaryotic expression systems, including human tumor cells, hen's eggs and plant cells, are now commonly used for production of protein pharmaceuticals. In some cases, these cells are engineered to contain specific processing enzymes, e.g. glycosyltransferases, for posttranslational modification of the protein. β-Glucosidase used for enzyme replacement therapy in Gaucher's disease (see Chap. 28) is produced in bioengineered Chinese hamster ovary (CHO) cells. The enzyme secreted from these cells contains a mannose-6-phosphate signal, so that it is taken up into lysosomes following intravenous injection. Humanized proteins may also be synthesized in murine cells, then processed by glycosidases and/or glycosyl transferases to yield a protein with the proper posttranslational modifications for use in human plasma or cells. In the not-to-distant future, it may be possible to by-pass all of these steps by gene therapy, i.e. by incorporating the gene of interest directly into the relevant human cells using viral vectors. Despite all these advances, the fundamental idea of a foreign piece of DNA being inserted into a particular restriction site of a plasmid or viral vector remains completely relevant today.
![]() Clinical box Production of recombinant proteins: insulin
Clinical box Production of recombinant proteins: insulin
A 13-year-old girl was admitted with dehydration, vomiting and weight loss. Her blood glucose level was 19.1 mmol/L(344 mg/dL) and she had ketonuria. A diagnosis of type 1 diabetes mellitus was made. She was started on recombinant human insulin, was rehydrated (Chapter 21), and made a prompt recovery.
Comment.
Prior to the advent of recombinant DNA technology, insulin therapy involved the use of animal insulins, most commonly pork or beef, which were chemically similar, but not identical, to human insulin. As a result of these differences, animal insulins often led to the development of antibodies, which reduced the efficacy of the insulin and could lead to treatment failures.
Insulin was the first clinically important human molecule to be produced by means of recombinant DNA technology. Following the cloning of the human insulin gene, large-scale production of pure human insulin was possible by inserting the cloned gene into a cell-based amplification system. Large amounts of insulin gene copies were produced, which were then expressed in either bacteria or yeast, and the resulting purified insulin was made available for use in treatment of diabetic patients. By this means, human recombinant insulin has largely replaced animal insulin in the treatment of diabetes. Other important recombinant human peptides used clinically include growth hormone, erythropoietin and parathyroid hormone.
![]() Advanced concept box Vector systems for cloning large DNA fragments
Advanced concept box Vector systems for cloning large DNA fragments
One critical factor in recombinant DNA manipulations is the size of the target DNA. Conventional bacterial plasmids, although convenient to work with, are limited in the size of insert they can accept; 1–2 kb is the common size of the insert, with an upper limit of 5-10 kb. Some modified plasmid vectors called cosmids can accept larger fragments up to 20 kb. Another commonly used vector that has the ability to accept larger DNA fragments is the bacteriophage lambda (λ). This viral particle contains a double-stranded DNA genome packaged within a protein coat. The λ-phage can infect E. coli cells with high efficiency and introduce its DNA into the bacterium. Infection leads to the replication of viral DNA and the synthesis of new viral particles, which can then lyse the host cell and infect neighboring cells to repeat the process. The viral DNA is then re-isolated to obtain the recombinant DNA.
Larger inserts can be cloned by using modified chromosomes from either bacteria (bacterial artificial chromosomes, BACs), or yeast (yeast artificial chromosomes, YACs). Such vectors can accommodate DNA fragments up to 1–2 Mb. BACs have been particularly important in putting together the sequence of the human genome.