ACUTE GLOMERULONEPHRITIS (GN)
Often called the “nephritic syndrome.” Characterized by development, over days, of azotemia, hypertension, edema, hematuria, proteinuria, and sometimes oliguria. Salt and water retention are due to reduced glomerular filtration rate (GFR) and may result in circulatory congestion. Red blood cell (RBC) casts on urinalysis confirm diagnosis (Dx). Proteinuria is usually <3 g/d. Most forms of acute GN are mediated by humoral immune mechanisms. Clinical course depends on underlying lesion (Table 152-1).
TABLE 152-1 CAUSES OF ACUTE GLOMERULONEPHRITIS

Acute Poststreptococcal GN
This is the prototype and most common cause in childhood. Nephritis develops 1–3 weeks after pharyngeal or cutaneous infection with “nephritogenic” strains of group A β-hemolytic streptococci. Dx depends on a positive pharyngeal or skin culture (if available), positive titers for antistreptococcal antigens (ASO, anti-DNAse, or antihyaluronidase), and hypocomplementemia. Renal biopsy reveals diffuse proliferative GN. Treatment consists of correction of fluid and electrolyte imbalance. In most cases the disease is self-limited, although the prognosis is less favorable and urinary abnormalities are more likely to persist in adults.
Postinfectious GN
May follow other bacterial, viral, and parasitic infections. Examples are bacterial endocarditis, sepsis, hepatitis B, and pneumococcal pneumonia. Features are milder than with poststreptococcal GN. Control of primary infection usually produces resolution of GN.
RAPIDLY PROGRESSIVE GLOMERULONEPHRITIS
Defined as a subacute reduction in GFR of >50%, with evidence of a proliferative GN; causes overlap with those of acute GN (Table 152-2). Broadly classified into three major subtypes on the basis of renal biopsy findings and pathophysiology: (1) immune complex–associated, e.g., in systemic lupus erythematosus (SLE); (2) “pauci-immune,” associated with antineutrophil cytoplasmic antibodies (ANCA); and (3) associated with anti–glomerular basement (anti-GBM) antibodies, e.g., in Goodpasture’s syndrome. All three forms will typically have a proliferative, crescentic GN by light microscopy but differ in the results of the immunofluorescence and electron microscopy components of the renal biopsy.
TABLE 152-2 CAUSES OF RAPIDLY PROGRESSIVE GLOMERULONEPHRITIS


SLE (Lupus)
Renal involvement is due to deposition of circulating immune complexes. Clinical features of SLE with or without renal involvement include arthralgias, “butterfly” skin rash, serositis, alopecia (hair loss), and central nervous system disease. Nephrotic syndrome with renal insufficiency is common. Renal biopsy reveals mesangial, focal, or diffuse GN and/or membranous nephropathy. Diffuse GN, the most common finding in renal biopsy series, is characterized by an active sediment, severe proteinuria, and progressive renal insufficiency and may have an ominous prognosis. Pts have a positive antinuclear antibody test, anti-dsDNA antibodies, and hypocomplementemia. Treatment includes glucocorticoids and cytotoxic agents. Oral or IV monthly cyclophosphamide is most commonly employed, typically for a period of 6 months; pts of childbearing age should first bank sperm and eggs. Mycophenolate mofetil is an alternative.
Antineutrophil Cytoplasmic Antibody (ANCA)–Associated, Pauci-Immune GN
May be renal-limited (idiopathic pauci-immune GN) or associated with systemic vasculitis [granulomatosis with polyangiitis (Wegener’s) or microscopic polyarteritis nodosa]. Defining characteristic is the presence of circulating ANCA. These are detected by immunofluorescence of alcohol-fixed neutrophils; a “perinuclear” pattern (pANCA) is usually due to antibodies against myeloperoxidase (MPO), whereas a “cytoplasmic” pattern (cANCA) is almost always due to reactivity against proteinase-3 (PR3). Confirmatory enzyme-linked immunosorbent assay testing against the MPO and PR3 antigens is mandatory, since the pANCA pattern can be caused by antibodies against other neutrophil components, e.g., lactoferrin; these do not have the same consistent relationship to vasculitis and pauci-immune GN. The anti-MPO or anti-PR3 titer does not always correlate with disease activity.
Pts typically have a prodromal, “flulike” syndrome, which may encompass myalgias, fever, arthralgias, anorexia, and weight loss. There may be associated cutaneous, pulmonary, upper respiratory (sinusitis), or neurologic (mononeuritis monoplex) complications of associated systemic vasculitis. In particular, pulmonary necrotizing capillaritis can lead to hemoptysis and pulmonary hemorrhage.
Standard initial therapy for ANCA-associated rapidly progressive GN includes methylprednisolone and cyclophosphamide; more specific depletion of B cells by anti-CD20 antibody therapy with rituximab is an alternative. Some centers will also utilize plasmapheresis in the initial management of pts with a severe pulmonary-renal syndrome or to stave off dialysis in pts with severe renal impairment. Steroids are quickly tapered soon after the acute inflammation subsides; cyclophosphamide is continued until a stable remission is achieved, typically within 3–6 months. Pts must receive prophylaxis for Pneumocystis carinii (jiroveci) pneumonia (PCP) with trimethoprim-sulfamethoxazole, atovaquone, or dapsone. Some form of maintenance immunosuppression is standard, typically for 12–18 months after achievement of a stable remission; drugs include methotrexate, mycophenolate mofetil, and azathioprine.
Anti–Glomerular Basement Membrane Disease
Caused by antibodies against the α3 NCI (noncollagenous) domain of type IV collagen; circulating anti-GBM antibody and linear immunofluorescence on renal biopsy establish the Dx. Pts may have isolated GN; Goodpasture’s syndrome encompasses GN and lung hemorrhage. Plasma exchange may produce remission; renal prognosis is worse in those who require dialytic support, with >50% crescents on renal biopsy, or creatinine >5–6 mg/dL. Severe lung hemorrhage is treated with IV glucocorticoids (e.g., 1 g/d × 3 days). Approximately 10–15% will also have ANCA against MPO, some with evidence of vasculitis, e.g., leukocytoclastic vasculitis in the skin.
Henoch-Schönlein Purpura
A generalized vasculitis causing IgA nephropathy, purpura, arthralgias, and abdominal pain; occurs mainly in children. Renal involvement is manifested by hematuria and proteinuria. Serum IgA is increased in half of pts. Renal biopsy is useful for prognosis. Treatment is symptomatic.
NEPHROTIC SYNDROME (NS)
Characterized by albuminuria (>3.5 g/d) and hypoalbuminemia (<30 g/L) and accompanied by edema, hyperlipidemia, and lipiduria. Protein excretion can be quantified by 24-h urine collection or by measurement of the urine protein:creatinine ratio or albumin:creatinine ratio on a random spot urine. The measurement of creatinine excretion helps define the adequacy of 24-h urine collections: daily creatinine excretion should be 20–25 mg/kg lean body weight in men and 15–20 mg/kg lean body weight in women. For random urine samples, the ratio of protein or albumin to creatinine in mg/dL approximates the 24-h urine protein excretion, since creatinine excretion is only slightly greater than 1000 mg/d per 1.73 m2. A urine protein:creatinine ratio of 5 is thus consistent with 5 g/d per 1.73 m2. Quantification of urine protein excretion on spot urines has largely supplanted formal 24-h urine collections, due to the greater ease and the need to verify a complete 24-h collection. The total protein:creatinine ratio does not detect microalbuminuria, a level of albumin excretion that is below the level of detection by tests for total protein; urine albumin: creatinine measurement is therefore preferred as a screening tool for lesser proteinuria.
In addition to edema, the complications of NS can include renal vein thrombosis and other thromboembolic events, infection, vitamin D deficiency, protein malnutrition, and drug toxicities due to decreased protein binding.
In adults, the most common cause of NS is diabetes. A minority of cases are secondary to SLE, amyloidosis, drugs, neoplasia, or other disorders (Table 152-3). By exclusion, the remainder are idiopathic. With the exception of diabetic nephropathy, a renal biopsy is required to make the diagnosis and determine therapy in NS.
TABLE 152-3 CAUSES OF NEPHROTIC SYNDROME (NS)

Minimal Change Disease
Causes about 10–15% of idiopathic NS in adults, but 70–90% of NS in children. Blood pressure is normal; GFR is normal or slightly reduced; urinary sediment is benign or may show few RBCs. Protein selectivity is variable in adults. Recent upper respiratory infection, allergies, or immunizations are present in some cases; nonsteroidal anti-inflammatory drugs can cause minimal change disease with interstitial nephritis. Acute renal failure may rarely occur, particularly among elderly persons. Renal biopsy shows only foot process fusion on electron microscopy. Remission of proteinuria with glucocorticoids carries a good prognosis; cytotoxic therapy may be required for relapse. Progression to renal failure is uncommon. Focal sclerosis has been suspected in some cases refractory to steroid therapy.
Membranous GN
Characterized by subepithelial IgG deposits; accounts for ~30% of idiopathic adult NS. Pts present with edema and nephrotic proteinuria. Blood pressure, GFR, and urine sediment are usually normal at initial presentation. Hypertension, mild renal insufficiency, and abnormal urine sediment develop later. Renal vein thrombosis is relatively common, more so than with other forms of NS. Underlying diseases such as SLE, hepatitis B, and solid tumors and exposure to such drugs as high-dose captopril or penicillamine should be sought. The majority of pts with idiopathic membranous GN have detectable circulating autoantibodies to the M-type phospholipase A2 (PLA2R), which is expressed in glomerular podocytes. Some pts progress to end-stage renal disease (ESRD); however, 20–33% may experience a spontaneous remission. Male gender, older age, hypertension, and persistence of significant proteinuria (>6 g/d) are associated with a higher risk of progressive disease. Optimal immunosuppressive therapy is controversial. Glucocorticoids alone are ineffective. Cytotoxic agents may promote complete or partial remission in some pts, as may cyclosporine. Anti-CD20 antibody therapy with rituximab has recently shown considerable promise, consistent with a role for B cells and anti-PLA2R antibodies in the pathophysiology. Reduction of proteinuria with angiotensin-converting enzyme (ACE) inhibitors and/or angiotensin receptor blockers (ARBs) is also an important mainstay of therapy.
Focal Glomerulosclerosis (FGS)
Can be primary or secondary. Primary tends to be more acute, similar to minimal change disease in abruptness of NS, but with added features of hypertension, renal insufficiency, and hematuria. Involves fibrosis of portions of some (primarily juxtamedullary) glomeruli and is found in ~35% of pts with NS. There are several different pathologic subtypes of idiopathic FGS, with prognostic implications. In particular, the “collapsing glomerulopathy” variant has pathologic similarity to HIV-associated nephropathy (HIVAN); both nephropathies cause rapidly progressive disease.
African Americans are disproportionately affected by FGS, HIVAN, and other nondiabetic renal disease, with higher incidence, greater susceptibility (HIVAN), and a much higher risk of developing ESRD. “African-specific” variants in the APOL1 gene, which encodes apolipoprotein L1 expressed in glomerular podocytes, have recently been implicated in this enhanced genetic risk.
Treatment of primary FGS typically begins with an extended course of steroids; fewer than half of pts undergo remission. Cyclosporine is an alternative therapy for maintenance of remission and for steroid-resistant pts. As in other glomerulopathies, reduction of proteinuria with ACE inhibitors and/or ARBs is also an important component of therapy. Finally, primary FGS may recur after renal transplant, when it may lead to loss of the allograft.
Secondary FGS can occur in the late stages of any form of kidney disease associated with nephron loss (e.g., remote GN, pyelonephritis, sickle cell disease, vesicoureteral reflux). Treatment includes anti-proteinuric therapy with ACE inhibition and blood pressure control. There is no benefit of glucocorticoids in secondary FGS. Clinical history, kidney size, biopsy findings, and associated conditions usually allow differentiation of primary versus secondary causes.
Membranoproliferative Glomerulonephritis (MPGN)
Mesangial expansion and proliferation extend into the capillary loop. Two ultrastructural variants exist. In MPGN I, subendothelial electron-dense deposits are present, C3 is deposited in a granular pattern indicative of immune-complex pathogenesis, and IgG and the early components of complement may or may not be present. In MPGN II, the lamina densa of the GBM is transformed into an electron-dense character, as is the basement membrane in Bowman’s capsule and tubules. C3 is found irregularly in the GBM. Small amounts of Ig (usually IgM) are present, but early components of complement are absent. Serum complement levels are decreased. MPGN affects young adults. Blood pressure and GFR are abnormal, and the urine sediment is active. Some have acute nephritis or hematuria. Similar lesions occur in SLE and hemolytic-uremic syndrome. Infection with hepatitis C virus (HCV) has been linked to MPGN, often with associated cryoglobulinemia. Treatment with interferon α and ribavirin has resulted in remission of renal disease in some cases, depending on HCV serotype; however, renal insufficiency typically precludes ribavirin therapy. Glucocorticoids, cytotoxic agents, antiplatelet agents, and plasmapheresis have been used with limited success; rituximab is a newer therapy with greater evident efficacy. MPGN may recur in allografts.
Diabetic Nephropathy
The most common cause of NS. Although prior duration of diabetes mellitus (DM) is variable, in type 1 DM proteinuria may develop 10–15 years after onset of diabetes, progress to NS, and then lead to renal failure over 3–5 years. Retinopathy is nearly universal in type 1 diabetics with nephropathy, so much so that the absence of retinopathy should prompt consideration of another glomerular lesion (e.g., membranous nephropathy). In contrast, only ~60% of type 2 diabetics with diabetic nephropathy have retinopathy. Clinical features include proteinuria, progressive hypertension, and progressive renal insufficiency. Pathologic changes include mesangial sclerosis, diffuse, and/or nodular (Kimmelstiel-Wilson) glomerulosclerosis. However, pts rarely undergo renal biopsy; to the extent that yearly measurement of microalbuminuria is routine management for all diabetics, the natural history is an important component of the diagnosis. Pts typically demonstrate progression from microalbuminuria (30–300 mg/24 h) to dipstick-positive proteinuria (>300 mg albuminuria) and then progressively overt proteinuria and chronic kidney disease. However, proteinuria can be quite variable in diabetic nephropathy, with as much as 25 g/24 h in the absence of profound renal insufficiency or alternatively with progressive renal insufficiency and stable, modest proteinuria.
Treatment with ACE inhibitors delays the onset of nephropathy and of ESRD in type 1 diabetics with microalbuminuria and/or declining renal function and should be instituted in all pts tolerant to that class of drug. If a cough develops in a pt treated with an ACE inhibitor, an ARB is the next best choice. Type 2 diabetics with microalbuminuria or proteinuria can be treated with ACE inhibitors or ARBs. Although long-term studies are lacking, many authorities advocate combining inhibitors of the renin-angiotensin-aldosterone system (RAAS), i.e., ARBs, ACE inhibitors, mineralocorticoid receptor blockers, and/or renin inhibitors, in pts with persistent, significant proteinuria. Hyperkalemia, hypotension, and/or worsening GFR can limit single or combined therapy with RAAS inhibitors. If hyperkalemia develops and cannot be controlled with (1) optimizing glucose control, (2) loop diuretics (if otherwise appropriate), or (3) treatment of metabolic acidosis (if present), then tight control of blood pressure with alternative agents is warranted.
Evaluation of NS is shown in Table 152-4.
TABLE 152-4 EVALUATION OF NEPHROTIC SYNDROME

ASYMPTOMATIC URINARY ABNORMALITIES
Proteinuria in the nonnephrotic range and/or hematuria unaccompanied by edema, reduced GFR, or hypertension can be due to multiple causes (Table 152-5).
TABLE 152-5 GLOMERULAR CAUSES OF ASYMPTOMATIC URINARY ABNORMALITIES


Thin Basement Membrane Nephropathy
Also known as benign familial hematuria, may cause up to 25% of isolated, sustained hematuria without proteinuria. Diffuse thinning of the glomerular basement membrane on renal biopsy, with minimal other changes. May be hereditary, caused in some instances by defects in type IV collagen. Pts have persistent glomerular hematuria, with minimal proteinuria. The renal prognosis is controversial but appears to be relatively benign.
IgA Nephropathy
Another very common cause of recurrent hematuria of glomerular origin; is most frequent in young men. Episodes of macroscopic hematuria are present with flulike symptoms, without skin rash, abdominal pain, or arthritis. Renal biopsy shows diffuse mesangial deposition of IgA, often with lesser amounts of IgG, nearly always by C3 and properdin but not by C1q or C4. Prognosis is variable; 50% develop ESRD within 25 years; men with hypertension and heavy proteinuria are at highest risk. Glucocorticoids and other immunosuppressive agents have not proved successful, except in pts who present with rapidly progressive GN. A randomized clinical trial of fish oil supplementation suggested a modest therapeutic benefit. Rarely recurs in allografts.
Glomerulopathies Associated with Multisystem Disease (Table 152-6)
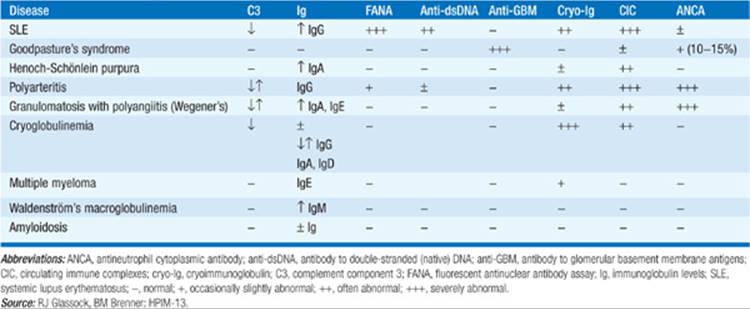
TABLE 152-6 SEROLOGIC FINDINGS IN SELECTED MULTISYSTEM DISEASES CAUSING GLOMERULAR DISEASE

For a more detailed discussion, see Lewis JB, Neilson EG: Glomerular Diseases, Chap. 283, p. 2334, in HPIM-18.