HEMOCHROMATOSIS
Hemochromatosis is a disorder of iron storage that results in increased intestinal iron absorption with Fe deposition and damage to many tissues. The classic clinical constellation of hemochromatosis is a pt presenting with bronze skin, liver disease, diabetes, arthropathy, cardiac conduction abnormalities, and hypogonadism. Two major causes of hemochromatosis exist: hereditary (due to inheritance of mutant HFE genes) and secondary iron overload (usually the result of ineffective erythropoiesis, such as thalassemia or sideroblastic anemia). HFE encodes a protein that is involved in cellular iron sensing and in regulating intestinal iron absorption. HFE mutations are very common in populations of Northern European origin (1 in 10 is a carrier). Heterozygotes are asymptomatic; homozygotes show a disease penetrance of ~30%. There is progressive iron overload, with clinical manifestations appearing after age 30–40, typically earlier in men than in women. Alcoholic liver disease and chronic excessive Fe ingestion may also be associated with a moderate increase in hepatic Fe and elevated body Fe stores.
Clinical Features
Early symptoms include weakness, lassitude, weight loss, a bronze pigmentation or darkening of skin, abdominal pain, and loss of libido. Hepatomegaly occurs in 95% of pts, sometimes in the presence of normal LFTs. If untreated, liver disease progresses to cirrhosis, and further to hepatocellular carcinoma in ~30% of pts with cirrhosis. Other manifestations include skin pigmentation (bronzing), diabetes mellitus (65% of pts), arthropathy (25–59%), cardiac arrhythmias and CHF (15%), and hypogonadotropic hypogonadism. Diabetes mellitus is more common in pts with a family history of diabetes, and hypogonadism may be an isolated early manifestation. Typical signs of portal hypertension and decompensated hepatic cirrhosis may appear late in the clinical course. Adrenal insufficiency, hypothyroidism, and hypoparathyroidism rarely occur.
Diagnosis
Serum Fe, percent transferrin saturation, and serum ferritin levels are increased. In an otherwise-healthy person, a fasting serum transferrin saturation >50% is abnormal and suggests homozygosity for hemochromatosis. In most untreated pts with hemochromatosis, the serum ferritin level is also greatly increased. If either the percent transferrin saturation or the serum ferritin level is abnormal, genetic testing for hemochromatosis should be performed. All first-degree relatives of pts with hemochromatosis should be tested for the C282Y and H63D mutations in HFE. Liver biopsy may be required in affected individuals to evaluate possible cirrhosis and to quantify tissue iron. An algorithm for evaluating pts with possible hemochromatosis is shown in Fig. 190-1. Death in untreated pts results from cardiac failure (30%), cirrhosis (25%), and hepatocellular carcinoma (30%); the latter may develop despite adequate Fe removal.
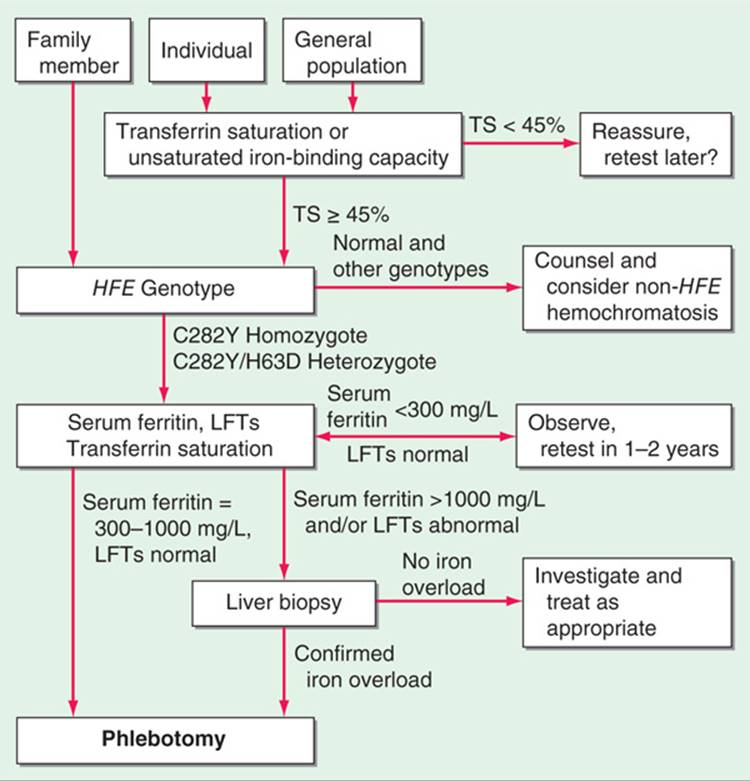
FIGURE 190-1 Algorithm for screening for HFE-associated hemochromatosis. LFT, liver function tests; TS, transferrin saturation. (From EJ Eijkelkamp et al: Can J Gastroenterol 14:2, 2000; with permission.)
TREATMENT Hemochromatosis
Therapy involves removal of excess body Fe, usually by intermittent phlebotomy, and supportive treatment of damaged organs. Since 1 unit of blood contains ~250 mg Fe, and since up to 25 g of Fe must be removed, phlebotomy is performed weekly for 1–2 years. Less frequent phlebotomy is then used to maintain serum Fe at 9–18 μmol/L (50–100 μg/dL). Chelating agents such as deferoxamine (infused subcutaneously using a portable pump) remove 10–20 mg iron per day, a fraction of that mobilized by weekly phlebotomy. Chelation therapy is indicated, however, when phlebotomy is inappropriate, such as with anemia or hypoproteinemia. Alcohol consumption should be eliminated. End-stage liver disease may require liver transplantation.
PORPHYRIAS
The porphyrias are inherited disturbances in heme biosynthesis. Each of the nine disorders causes a unique pattern of overproduction, accumulation, and excretion of intermediates of heme synthesis. These disorders are classified as either hepatic or erythropoietic, depending on the primary site of overproduction and accumulation of the porphyrin precursor or porphyrin. The major manifestations of the hepatic porphyrias are neurologic (neuropathic abdominal pain, neuropathy, and mental disturbances), whereas the erythropoietic porphyrias characteristically cause cutaneous photosensitivity. Laboratory testing is required to confirm or exclude the various types of porphyria. However, a definite diagnosis requires demonstration of the specific enzyme deficiency or gene defect. Only the three most common porphyrias are discussed here.
ACUTE INTERMITTENT PORPHYRIA
This is an autosomal dominant disorder with variable expressivity caused by partial (50%) deficiency in hydroxymethylbilane synthase. It has a prevalence of 1–3 in 100,000 but is much more common in certain parts of the world (Northern Sweden, Great Britain). Manifestations include colicky abdominal pain, vomiting, constipation, port wine–colored urine, and neurologic and psychiatric disturbances. Acute attacks rarely occur before puberty and may last from days to months. Photosensitivity does not occur. Clinical and biochemical manifestations may be precipitated by barbiturates, anticonvulsants, estrogens, oral contraceptives, the luteal phase of the menstrual cycle, alcohol, or low-calorie diets. Diagnosis is established by demonstrating elevation of urinary porphobilinogen (PBG) and γ-aminolevulinic acid (ALA) during an acute attack.
TREATMENT Acute Intermittent Porphyria
As soon as possible after the onset of an attack, 3–4 mg of heme, in the form of heme arginate, heme albumin, or hematin, should be infused daily for 4 days. Heme acts by inhibiting ALA synthase, thereby restraining ALA and PBG production. Administration of IV glucose at rates up to 20 g/h or parenteral nutrition, if oral feeding is not possible for long periods, can be effective in acute attacks. Narcotic analgesics may be required during acute attacks for abdominal pain, and phenothiazines are useful for nausea, vomiting, anxiety, and restlessness. Treatment between attacks involves adequate nutritional intake, avoidance of drugs known to exacerbate the disease, and prompt treatment of other inter-current diseases or infections.
PORPHYRIA CUTANEA TARDA
This is the most common porphyria (2–4 in 100,000) and is characterized by cutaneous photosensitivity and, usually, hepatic disease. It is due to partial deficiency (familial, sporadic, or acquired) of hepatic uroporphyrinogen decarboxylase. Photosensitivity causes facial pigmentation, increased fragility of skin, erythema, and vesicular and ulcerative lesions, typically involving face, forehead, and forearms. Neurologic manifestations are not observed. Contributing factors include excess alcohol, iron, and estrogens. Pts with liver disease are at risk for cirrhosis and hepatocellular carcinoma. Plasma and urine uroporphyrin and 7-carboxylate porphyrin are increased.
TREATMENT Porphyria Cutanea Tarda
Avoidance of precipitating factors, including abstinence from alcohol, estrogens, iron supplements, and other exacerbating drugs, is the first line of therapy. A complete response can almost always be achieved by repeated phlebotomy (every 1–2 weeks) until hepatic iron is reduced. Chloroquine or hydroxychloroquine may be used in low doses (e.g., 125 mg chloroquine phosphate twice weekly) to promote porphyrin excretion in pts unable to undergo or unresponsive to phlebotomy.
ERYTHROPOIETIC PROTOPORPHYRIA
Erythropoietic protoporphyria is an autosomal dominant disorder due to partial deficiency of ferrochelatase, the last enzyme in the heme biosynthetic pathway. Its prevalence is 1 in 100,000. Porphyrins (primarily protoporphyrin IX) from bone marrow erythrocytes and plasma are deposited in the skin and lead to cutaneous photosensitivity. Skin photosensitivity usually begins in childhood. The skin manifestations differ from those of other porphyrias, in that vesicular lesions are uncommon. Redness, swelling, burning, and itching can develop within minutes of sun exposure and resemble angioedema. Symptoms may seem out of proportion to the visible skin lesions. Chronic skin changes may include lichenification, leathery pseudovesicles, labial grooving, and nail changes. Liver function is usually normal, but liver disease and gallstones may occur. Protoporphyrin levels are increased in bone marrow, circulating erythrocytes, plasma, bile, and feces; protoporphyrin in erythrocytes is free rather than zinc-complexed as it is in other types of porphyria or hematologic disorders. Urinary porphyrin levels are normal. Diagnosis is confirmed by identifying a mutation in the ferrochelatase gene.
TREATMENT Erythropoietic Protoporphyria
Avoidance of sun exposure is essential. Oral β-carotene (120–180 mg/d) improves tolerance to sunlight in many pts. The dosage may be adjusted to maintain serum carotene levels between 10 and 15 μmol/L (600–800 μg/dL). Cholestyramine or activated charcoal may promote fecal excretion of protoporphyrin. Plasmapheresis or IV heme therapy may be beneficial.
WILSON’S DISEASE
Wilson’s disease is a rare inherited disorder of copper metabolism, resulting in the toxic accumulation of copper in the liver, brain, and other organs. Individuals with Wilson’s disease have mutations in the ATP7B gene, which encodes a membrane-bound copper-transporting ATPase. Deficiency of this protein impairs copper excretion into the bile and copper incorporation into ceruloplasmin, leading to its rapid degradation.
Clinical Features
Clinical manifestations typically appear in the mid- to late-teen years but may occur later. Hepatic disease may present as hepatitis, cirrhosis, or hepatic decompensation. In other pts, neurologic or psychiatric disturbances are the first clinical sign and are always accompanied by Kayser-Fleischer rings (corneal deposits of copper). Dystonia, incoordination, or tremor may be present, and dysarthria and dysphagia are common. Autonomic disturbances also may be present. Microscopic hematuria is common. In about 5% of pts, the first manifestation may be primary or secondary amenorrhea or repeated spontaneous abortions.
Diagnosis
Serum ceruloplasmin levels are often low but may be normal in up to 10% of pts. Urine copper levels are elevated. The “gold standard” for diagnosis is an elevated copper level on liver biopsy. Genetic testing is not currently practical because of the large number of mutations that can be responsible.
TREATMENT Wilson’s Disease
Hepatitis or cirrhosis without decompensation should be treated with zinc acetate (50 mg elemental Zn PO three times a day). Zinc is effective by blocking intestinal copper absorption and inducing metallothionein, which sequesters copper in an nontoxic complex. For pts with hepatic decompensation, the chelator trientene (500 mg PO twice a day) plus zinc (separated by at least 1 h to avoid zinc chelation in the intestinal lumen) is recommended, although liver transplantation should be considered for severe hepatic decompensation. For initial neurologic therapy, trientine and zinc are recommended for 8 weeks, followed by therapy with zinc alone. Tetrathiomolybdate is an alternative therapeutic option available in the future. Penicillamine is no longer first-line therapy. Zinc treatment does not require monitoring for toxicity, and 24-h urine copper can be followed for a therapeutic response. Trientine may induce bone marrow suppression and proteinuria. With chelation therapy, measuring free serum copper levels (adjusting total serum copper for ceruloplasmin copper) rather than urine copper is used to monitor therapeutic response. Anticopper therapy must be lifelong.

For a more detailed discussion, see Powell LW: Hemochromatosis, Chap. 357, p. 3162; Desnick RJ, Balwani M: The Porphyrias, Chap. 358, p. 3167; and Brewer GJ: Wilson’s Disease, Chap. 360, p. 3188, in HPIM-18.