Andreas Greinacher1 & Theodore E. Warkentin2
1Department of Immunology and Transfusion Medicine, Universitätsmedizin Greifswald, Greifswald, Germany
2Department of Pathology and Molecular Medicine and Department of Medicine, Michael G. DeGroote School of Medicine, McMaster University, Transfusion Medicine, Hamilton Regional Laboratory Medicine Program and Service of Clinical Hematology, Hamilton Health Sciences, Hamilton, Ontario, Canada
Heparin-induced thrombocytopenia (HIT) is an antibody-mediated adverse effect of heparin. It is highly prothrombotic and treatment usually requires substitution of heparin with a rapidly acting nonheparin anticoagulant; vitamin K antagonists (warfarin) are contraindicated during the acute phase of HIT because their use can precipitate limb necrosis due to microthrombosis. Prophylactic platelet transfusions should be minimized. Given these special treatment considerations, the challenge is to distinguish HIT from non-HIT thrombocytopenia. Management of HIT requires knowledge in immunohaematology and haemostasis. Table 30.1 lists features of HIT with particular relevance to the transfusion medicine specialist.
Table 30.1 HIT issues relevant to transfusion medicine.
|
HIT-related item |
Transfusion medicine-related comment |
|
PF4/heparin complexes form at optimal stoichiometric ratio |
The Coombs test requires an optimal concentration of the antihuman immunoglobulin antibody to achieve agglutination of red cells |
|
Acute HIT activates platelets, monocytes, endothelial cells and the coagulation cascade |
Acute haemolytic transfusion reaction activates platelets, leucocytes, endothelial cells and the clotting cascade |
|
Typical-onset HIT (day 5–14) |
Timing resembles that of delayed haemolytic transfusion reaction |
|
Rapid-onset HIT (<1 day) |
Timing resembles that of acute haemolytic transfusion reaction (i.e. due to pre-existing anti-red-cell alloantibodies) |
|
‘Delayed-onset’ HIT antibodies bind to and activate platelets even in the absence of heparin |
In posttransfusion purpura, alloantibodies boosted by transfusing HPA-1a positive platelets bind to the patient's own (HPA-1a negative) platelets, causing severe thrombocytopenia (see Chapter 12) |
|
Functional (platelet activation) assays are more predictive for HIT than immunoassays |
HLA antibodies that test positive in lymphocytotoxicity tests are more clinically relevant compared to ELISA-only reactive HLA antibodies |
|
Most ELISA-positive patients do not develop HIT |
Most anti-red-blood-cell alloantibodies do not cause intravascular haemolysis |
|
Particle gel immunoassay |
Rapid assay utilizing gel card technology commonly used in transfusion medicine |
|
Platelet transfusions (prophylactic) |
Relatively contraindicated in HIT |
|
PCCs contain heparin |
PCCs are relatively contraindicated during acute HIT |
|
High-dose intravenous IgG (IVIgG) |
IVIgG is occasionally used as adjunctive treatment for severe HIT |
|
ELISA, enzyme-linked immunosorbert assay; PCC, prothrombin complex concentrates; HLA, human leucocyte antigen; PF4, platelet factor 4. |
|
Pathogenesis
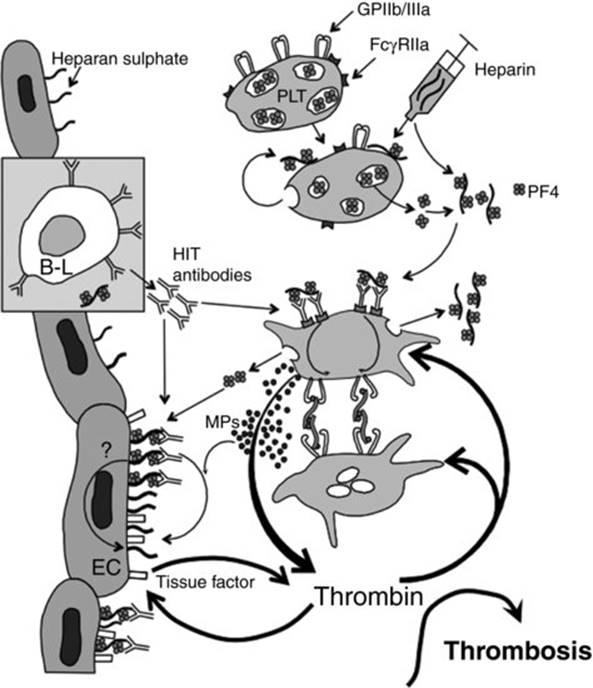
Figure 30.1 illustrates the pathogenesis of HIT [1]. Key features include:
· Antigens form when platelet factor 4 (PF4) – a positively charged 31 kDa tetrameric member of the C-X-C subfamily of chemokines – forms multimolecular complexes with (negatively charged) heparin when both are present at stoichiometrically optimal concentrations (1:1 to 2:1 ratio of PF4 : heparin).
· Both PF4 and heparin bind to platelet surfaces; thus, in situ formation of PF4/heparin complexes on platelet membranes localizes subsequent formation of PF4/heparin/IgG immune complexes also to the platelet surfaces; i.e. there are no circulating immune complexes in HIT.
· The HIT antigen(s) reside(s) on PF4, rather than on heparin; indeed, nonheparin polyanions (e.g. polyvinyl sulfonate, or PVS) can substitute for heparin in forming HIT antigens.
· Ultralarge PF4/heparin complexes are more readily formed with unfractionated heparin (UFH) compared with low-molecular-weight heparin (LMWH), perhaps explaining the tenfold greater risk of HIT with UFH versus LMWH.
· Heparin causes platelet activation and release of PF4. However, immunization occurs most often post-surgery and major trauma (perhaps reflecting PF4 release from activated platelets and/or proinflammatory factors).
· Anti-PF4/heparin antibodies become detectable ∼4 days (median) after an immunizing heparin exposure, with detection of platelet-activating antibodies 1 or 2 days later [2,3].
· Anti-PF4/heparin antibodies of IgG and/or IgA and/or IgM can be formed (relative frequency, IgG > IgA > IgM); however, only IgG antibodies have the potential to cause HIT, because platelet activation occurs only when multimolecular complexes of PF4/heparin/IgG result in clustering of the platelet Fc receptors (FcγIIa), causing intravascular platelet activation.
· HIT does not exhibit features of a classic primary immune response: even when HIT occurs during a patient's very first exposure to heparin, IgG antibodies are readily detected after only 4 to 5 days, whereas IgM antibodies usually are not detected. If IgM antibodies are found, they become detectable at the same time as IgG (i.e. no IgM precedence).
· These atypical features of the HIT immune response could reflect presensitization due to exposure to bacteria, as negatively charged molecules on bacterial surfaces bind PF4 in a way that exposes HIT antigens [4].
· Platelet activation in HIT includes formation of procoagulant platelet-derived microparticles.
· Other procoagulant features of HIT include monocyte and endothelial cell activation, and neutralization of heparin by PF4.
· Sometimes, HIT antibodies strongly activate platelets in the absence of pharmacologic heparin (heparin-‘independent' platelet activation): this is a feature of ‘delayed-onset’ HIT [5].
Fig 30.1 Pathogenesis of HIT. Platelet activation, either via binding of heparin to platelets (PLT) or by other mechanisms (e.g. surgery), leads to release of platelet factor 4 (PF4) from platelet α-granules. PF4/heparin complexes form, which in some patients triggers generation of platelet-activating anti-PF4/heparin antibodies (‘HIT antibodies’), predominantly of the IgG class. Multimolecular complexes comprised of PF4, heparin and IgG are formed on platelet surfaces, leading to crosslinking of the platelet Fc receptors (FcγRIIa). This produces potent platelet activation, including: conformational changes in the platelet fibrinogen receptors (GPIIb/IIIa), resulting in platelet aggregation; procoagulant changes in the platelet surface – including generation of procoagulant, platelet-derived microparticles (MPs) – leading to thrombin generation; and further release of granule constituents such as PF4, triggering even more IgG-mediated platelet activation. Further, PF4 binds to endothelial cell (EC) heparan sulfate, resulting in HIT antibody binding to endothelial PF4/heparin complexes and, possibly, EC activation and expression of endothelial tissue factor (open rectangle), contributing further to thrombin generation. Thrombin activates platelets and endothelium, leading to thrombosis. Reprinted from Warkentin et al. [1], with modifications, with permission.
Epidemiology
· The overall frequency of HIT among heparin-exposed inpatients is ∼0.2%.
· The frequency of HIT approaches 5–10% when there are multiple concurrent risk factors for HIT, e.g. (a) UFH use (versus LMWH or fondaparinux) for (b) at least 10–14 days (when antibodies peak), (c) postorthopedic surgery and (d) female sex (1.5–2.0×greater risk of HIT in females versus males) [6].
· HIT occurs more often in post-surgery patients than in medical patients [6]. HIT is rare in pregnancy and in paediatric patients, and probably does not occur in neonates.
· UFH is rarely administered nowadays to post-orthopaedic surgery patients. Thus, HIT nowadays occurs most often in postcardiac/postvascular surgery patients and general surgery patients who receive postoperative UFH thromboprophylaxis.
· Rarely, a transient HIT-mimicking syndrome with thrombocytopenia, thrombosis and high levels of platelet-activating anti-PF4/heparin antibodies can occur without proximate exposure to heparin, but after infection or surgery (‘spontaneous HIT’) [7].
HIT: a ‘clinicopathologic’ syndrome
· Table 30.2 summarizes the major clinical and laboratory features of HIT [8].
Fig 30.2 Iceberg model using published data [10]. The central ‘iceberg’ depicts three different antibody reaction profiles, as defined by a platelet activation test (HIPA) and two ELISAs (IgG-ELISA, poly-ELISA). The table on the far left shows the pretest probability scores (4Ts) for the three different antibody reaction profiles, as well as for patients who test negative in both ELISAs (bottom row of 4Ts table). On the far right, the corresponding results in the particle gel immunoassay (PaGIA) are shown. The data demonstrate that the sensitivity of the PaGIA for definite HIT (35 patients depicted as the ‘tip of the iceberg’) is only 94% (33/35). At the other extreme, ∼5% of the patients who have no antibodies by ELISA will test positive in the PaGIA (17/376). ELISA (or EIA), enzyme-linked immunosorbent assay (or enzyme-immunoassay); HIT, heparin-induced thrombocytopenia; PaGIA, particle gel immunoassay. Reprinted, with permission, from Warkentin & Linkins [9].
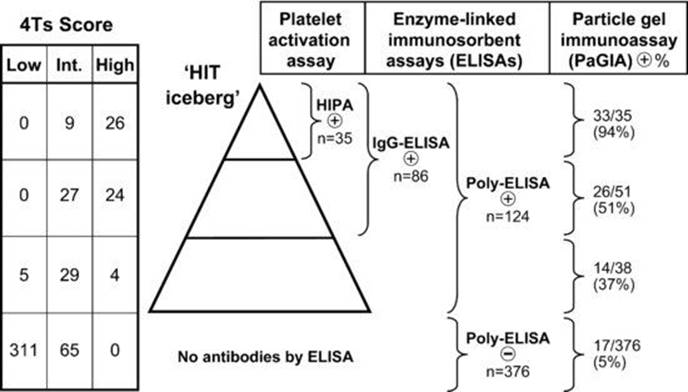
Table 30.2 HIT viewed as a clinical–pathological syndrome.
|
Clinical |
Pathological |
|
At least one of: · Thrombocytopenia · Thrombosis (e.g. venous: DVT, pulmonary embolism, venous limb gangrene, adrenal haemorrhage, cerebral vein thrombosis, splanchnic vein thrombosis; arterial: limb artery thrombosis, stroke, myocardial infarction, mesenteric artery thrombosis, miscellaneous artery; microvascular) · Necrotizing skin lesions at heparin injection sites · Acute anaphylactoid reactions · Disseminated intravascular coagulation (DIC)
|
Heparin-dependent, platelet-activating IgG · Positive platelet activation assay (e.g. SRA, HIPA) · Positive anti-PF4/polyanion-IgG ELISA (infers possible presence of platelet-activating IgG |
|
DVT, deep-vein thrombosis; ELISA, enzyme-linked immunosorbent assay; HIPA, heparin-induced platelet activation (test); PF4, platelet factor 4; SRA, serotonin-release assay. |
|
Iceberg model (Figure 30.2)
· HIT occurs in a minority of patients who form anti-PF4/heparin antibodies; e.g. anti-PF4/heparin antibodies are detectable in 50–80% of postcardiac surgery patients, yet HIT occurs in only 1–2% of these patients.
· According to the ‘iceberg model’, HIT occurs in the subset of patients who form strong heparin-dependent, platelet-activating antibodies of the IgG class; such antibodies are also readily detectable by PF4-dependent enzyme-linked immunosorbent assay (ELISA) [9,10].
· Diagnostic sensitivity of the three major types of assays – ELISA-IgG/A/M, ELISA-IgG, washed platelet activation assay – are similarly high (>99%); however, their diagnostic specificity varies, as follows: platelet activation assays > ELISA-IgG > ELISA-IgG/A/M.
Clinical picture
Thrombocytopenia
· HIT usually results in mild-to-moderate thrombocytopenia (median platelet count nadir, 60×109/L (∼90% have a nadir between 15 and 150×109/L) [11].
· In >90% of patients, the platelet count falls by >50% from the peak platelet count that immediately precedes the HIT-associated platelet count fall.
Timing
· ‘Typical-onset’ HIT indicates thrombocytopenia that begins 5–10 days after an immunizing heparin exposure [12].
· ‘Rapid-onset’ HIT refers to a platelet count fall that begins abruptly (<24 hours) after administration of heparin or a dose increase. Almost invariably, patients have been exposed to heparin within the recent past (past 5–100 days) [12].
· HIT antibodies are remarkably transient, becoming undetectable at a median of 50 to 85 days (depending on the assay performed) after an episode of HIT [12]. Antibodies have been reported to become substantially weaker within a week – with platelet count recovery – even if heparin is continued [2]. This indicates that a subclass of antibody-producing B cells is involved in HIT, which differs from classic immunohaematologic responses against alloantigens.
· ‘Delayed-onset’ HIT denotes thrombocytopenia that begins after the immunizing heparin exposure has been stopped or that worsens after stopping heparin; patient serum activates platelets in vitro even in the absence of pharmacologic heparin (heparin-‘independent' platelet activation) and have strongly positive ELISAs [5]. Such patients often have disseminated intravascular coagulation (DIC). The disorder resembles a transient autoimmune reaction.
· ‘Persisting’ HIT refers to thrombocytopenia that is slow to recover (∼1% of HIT patients take >1 month for the platelet count to rise to >150 × 109/L. In these patients, platelet numbers increase in parallel with gradually declining levels of heparin-independent platelet-activating antibodies.
Thrombosis and other sequelae
· HIT is strongly associated with venous and/or arterial thrombosis (relative risk, 10 to 15) [11].
· Thrombosis risk parallels the degree of thrombocytopenia, ranging from ∼50% for patients with mild thrombocytopenia (∼150×109/L) to ∼90% for patients with severe thrombocytopenia (∼20×109/L).
· Limb loss occurs in ∼5% of patients with HIT: explanations include limb arterial thrombosis, warfarin-induced venous limb gangrene [13] and DIC-associated microvascular thrombosis.
· Venous limb gangrene is acral (distal extremity) necrosis in a limb with deep-vein thrombosis (DVT) that occurs despite palpable or Doppler-identifiable arterial pulses. Patients usually have a supratherapeutic INR (>3.5) as a result of anticoagulation with a vitamin K antagonist. A prodromal state is phlegmasia cerulea dolens, i.e. an inflamed, ischaemic, painful limb.
· Venous predominates over arterial thrombosis (∼4:1 ratio) [11], except in patients with arteriopathy (∼1:1 ratio in postcardiac/postvascular surgery patients).
· Venous thrombotic events include (listed in descending order of frequency): venous thromboembolism (DVT > pulmonary embolism) > adrenal vein thrombosis > cerebral venous (dural sinus) thrombosis > splanchnic vein thrombosis.
· Adrenal vein thrombosis presents as unilateral or bilateral adrenal haemorrhage; when bilateral, death due to acute adrenal failure can occur (special relevance for critically ill patients).
· Arterial thrombotic events include: limb artery thrombosis > cerebral artery thrombosis > myocardial infarction.
· Overt (decompensated) DIC occurs in 10–15% of patients with HIT, usually with platelet count nadirs < 20×109/L; laboratory features include relative/absolute hypofibrinogenemia, elevated international normalized ratio (INR) and/or activated partial thromboplastin time (APTT) and (rarely) microangiopathy (red cell fragments, elevated lactate dehydrogenase, circulating normoblasts). Clinical features include microvascular thrombosis (e.g. ischaemic limb necrosis despite palpable pulses) and increased risk of treatment failure due to APTT confounding (discussed subsequently).
Anaphylactoid reactions occur in ∼25% of HIT patients who receive an intravenous UFH bolus and occasionally in patients administered subcutaneous LMWH (Table 30.3). There is an associated abrupt decrease in platelet count that can recover quickly after stopping heparin.
Table 30.3 Anaphylactoid reactions associated with acute (rapid-onset) HIT.
|
Timing: onset 5–30 minutes after intravenous unfractionated heparin bolus (less commonly, following intravenous or subcutaneous low-molecular-weight heparin administration) |
|
Clinical context: recent use of heparin (past 7–100 days) |
|
Laboratory features: abrupt, sometimes rapidly reversible fall in the platelet count |
|
Signs and symptoms: |
|
Inflammatory: chills, rigors, fever and flushing |
|
Cardiorespiratory: tachycardia, hypertension, |
|
Gastrointestinal: nausea, vomiting and large-volume |
|
Neurological: pounding headache, transient global |
Pretest probability scores
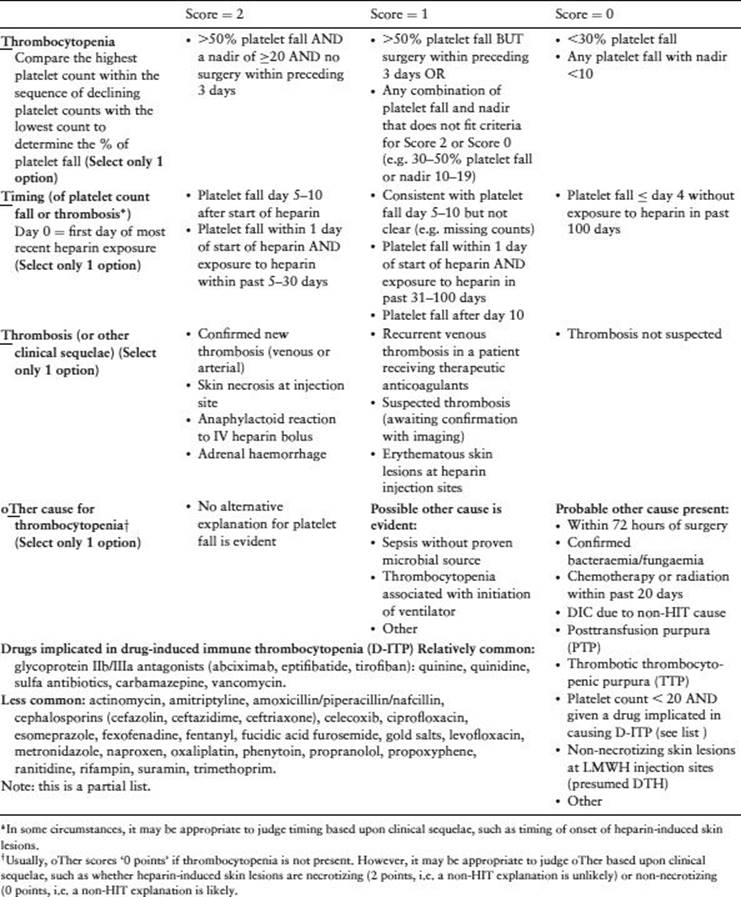
· The ‘4Ts’ is a pretest probability score that estimates the likelihood of HIT based upon: thrombocytopenia, timing (of platelet count fall or thrombosis), thrombosis (or other sequelae of HIT) and other causes for thrombocytopenia (Table 30.4) [14]. Low scores (3 or fewer points) are associated with <2% probability of HIT, whereas high scores (6 to 8 points) indicate ∼50% frequency of HIT.
· A more recent scoring system is the HIT expert probability (HEP) score, which requires further validation; like the 4Ts system, the HEP score evaluates the extent and timing of thrombocytopenia, the presence of thrombosis (or other HIT sequelae) and other potential explanations for thrombocytopenia, but assigns different numerical scores.
· Pretest probability scores are especially useful if interpreted in combination with certain immunoassays so as to predict the posttest likelihood of HIT.
· Critically ill patients with low pretest probability scores may not have HIT even if they test positive [15].
Table 30.4 The 4Ts pretest probability score. Reprinted, with permission, from Warkentin & Linkins [14].
Laboratory testing
Two general types of assays detect HIT antibodies: (a) platelet activation (or functional) assays and (b) PF4-dependent immunoassays.
· An unusual feature of HIT is that patient serum/ plasma-based assays are very sensitive for detecting HIT antibodies, even at the earliest phase of the platelet count decline [3]. (In contrast, sensitivity is lower for detecting circulating alloantibodies in delayed haemolytic transfusion reactions if antigen-positive red cells remain in circulation; in this situation, the direct Coombs test is more sensitive.)
· A characteristic feature is inhibition of reactivity at very high concentrations of UFH (10–100 U/mL), due to disruption of antigenic PF4/heparin complexes.
· In the absence of new clinical events (e.g. new thrombosis, new platelet count fall), a negative assay for HIT antibodies should not be automatically repeated a few days later; this is because a subsequent positive test result is much more likely to indicate subclinical seroconversion than ‘true’ HIT [3].
Platelet activation assays
Washed platelet activation assays
· The best operating characteristics (highest sensitivity-specificity trade-off) are seen with the washed platelet activation assays, the 14Cserotonin-release assay (SRA) and the heparin-induced platelet activation (HIPA) test.
· The SRA is performed in North America, using well-characterized (pedigree) donors, whereas the HIPA test is more widely used in Europe, and is usually performed with (random) donors at blood donation centres (four donors are tested separately to compensate for variable donor-dependent reactivity to HIT sera).
· Quality control manoeuvres include use of: (a) negative and graded (including weak-positive HIT serum) controls, (b) Fc receptor-blocking monoclonal antibodies (to confirm platelet activation occurs through platelet Fc receptors and (c) parallel testing in a PF4-dependent ELISA (expected to be positive if the SRA or HIPA is positive – per iceberg model).
Other platelet aggregation assays
· Standard platelet aggregometry (using patient platelet-poor plasma tested against normal donor platelet-rich plasma) is not recommended, due to suboptimal sensitivity and specificity and low test/control sample throughput.
· A whole blood aggregometry assay (Multiplate®) seems to have comparable sensitivity to washed platelet assays for detecting platelet-activating HIT antibodies if a highly reactive donor is used.
Table 30.5 PF4-dependent antigen assays (immunoassays).
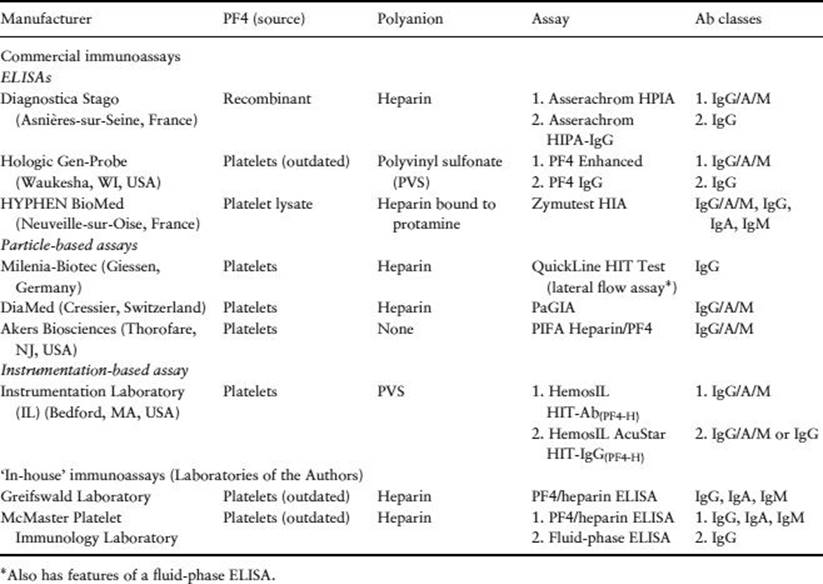
PF4-dependent immunoassays (antigen assays) (Table 30.5)
Enzyme-linked immunosorbent assays (solid-phase assays)
· Three commercial ELISAs are available to detect anti-PF4/heparin antibodies; reference centres also offer in-house assays. ELISAs are currently the most widely used tests for HIT.
· ‘Polyspecific’ ELISAs detect antibodies of the three major immunoglobulin classes (IgG/A/M).
· ‘IgG-specific’ ELISAs are preferred because their sensitivity is similarly high as the polyspecific assays, with substantially greater diagnostic specificity [9,10].
· The magnitude of a positive ELISA result, expressed in optical density (OD) units, predicts for greater likelihood of a positive platelet activation test. For an ELISA with a positive OD range of 0.40 to 3.00 OD units, approximate frequencies of positive activation assays are [16]:
0.40 to 1.00, ∼5%
1.00 to 1.50, ∼20%
1.50 to 2.00, ∼50%
>2.00, ∼90%
· Diagnostic specificity is enhanced somewhat using a high heparin confirmatory step, especially at weak-positive OD values (0.40 to 1.00). However, at higher OD values, lack of high heparin inhibition does not necessarily rule out platelet-activating HIT antibodies.
Fluid-phase immunoassays
Two fluid-phase immunoassays have been described; these avoid denaturation of PF4-dependent antigens (as can occur in solid-phase ELISAs), potentially increasing diagnostic specificity.
· Sepharose G fluid-phase (IgG-specific) ELISA (in-house assay). After binding of antibodies to (5% biotinylated) PF4 in the fluid phase, IgG antibodies are captured using Sepharose G. After washing, the amount of biotin-PF4/heparin-antibody complexes immobilized to the beads is measured using peroxidase substrate after initial incubation with streptavidin-conjugated peroxidase.
· Gold nanoparticle-based fluid-phase ELISA (rapid assay). In this ‘lateral-flow immunoassay’, capillary action causes the test sample to interact sequentially with antigen (ligand-labelled PF4/polyanion complexes), then with (red-coloured) gold nanoparticles coated with antiligand and then with immobilized goat antihuman IgG. A positive reaction is a bold-coloured line, which can be read visually or quantitatively with an automated reader. The turnaround time is only 15 minutes after preparation of serum, and the single-assay design facilitates on-demand testing.
Particle-based solid-phase immunoassays (rapid assays)
· Particle gel immunoassay (PaGIA). This assay utilizes a gel centrifugation technology system widely used in transfusion medicine. The manufacturer has prepared red, high-density polystyrene beads to which PF4/heparin complexes have been bound. After addition of patient serum/plasma, anti-PF4/heparin antibodies (if present) bind to the antigen-coated beads; a secondary antihuman immunoglobulin antibody is added into the sephacryl gel. Upon centrifugation, agglutinated beads (indicating the presence of anti-PF4/heparin antibodies) do not migrate through the sephacryl gel, whereas nonagglutinated beads (indicating the absence of antibodies) pass through the gel, forming a red band at the bottom. Sensitivity is lower than with the ELISAs (∼90–95% versus ∼99%) (Figure 30.2). The diagnostic specificity is intermediate between that of the (washed) platelet activation assay and ELISA. A positive reaction at 1/4 dilution of patient serum/plasma is more specific for HIT and a positive reaction at 1/32 dilution or greater predicts the presence of platelet-activating antibodies [17].
· Particle immunofiltration assay (PIFA). This assay utilizes a PIFA system, wherein patient serum is added to a reaction well containing dyed particles coated with PF4 (not PF4/heparin). Subsequently, nonagglutinated – but not agglutinated particles – will migrate through the membrane filter. Thus, a negative test is shown by a blue colour in the result well, whereas no colour indicates a positive test. The assay performed poorly in two reference laboratories and its use is not recommended.
Instrumentation-based immunoassays
Two automated assays that utilize proprietary instruments have recently been developed.
· HemosIL HIT-Ab(PF4-H). Using an analyser of the ACL TOP® family, this is a latex particle enhanced immunoturbidimetric assay that detects anti-PF4/heparin antibodies of all classes. In this competitive agglutination assay, the presence of anti-PF4/heparin antibodies within the patient sample will inhibit the binding of an HIT-mimicking monoclonal antibody (bound to latex particles) against PF4/PVS in solution. The degree of agglutination is inversely proportional to the level of anti-PF4/heparin antibodies (assessed by a decrease in light transmittance). A positive sample will therefore produce a lower OD than the negative control samples (the software automatically reports the results in U/mL as the inverse proportion). A positive test is a result ≥1.0 U/mL. The technology allows for rapid, on-demand single-patient testing.
· HemosIL AcuStar HIT-IgG(PF4-H). Using an ACL AcuStar® system instrument, this is a chemiluminescence assay that is also based upon binding of anti-PF4/heparin antibodies within patient serum/plasma to PF4/PVS. Magnetic particles coated with PF4/PVS capture anti-PF4/heparin antibodies present within a patient sample. After incubation, magnetic separation and a wash step, a tracer consisting of an isoluminol-labelled antihuman IgG antibody (or a mixture of three isoluminol-labelled monoclonal antibodies [anti-IgG/A/M]) is added, which binds to the captured anti-PF4/heparin antibodies on the particles. After a second incubation, magnetic separation and a wash step, reagents that trigger the luminescent reaction are added and the emitted light is measured as relative light units (RLUs) by the instrument's optical system. The RLUs are directly proportional to anti-PF4/heparin antibody concentrations. Like the ELISAs, higher assay results indicate a greater likelihood of HIT.
Treatment
Treatment principles
The treatment principles of strongly suspected or confirmed HIT are [18]:
· Substitute heparin with a rapidly acting nonheparin anticoagulant, usually in therapeutic doses.
· Avoid/postpone warfarin pending platelet count recovery.
· Minimize prophylactic platelet transfusions.
· Test for HIT antibodies.
· Investigate for lower-limb DVT (e.g. ultrasound), even if not clinically apparent.
Rapidly acting, nonheparin anticoagulants
· Anticoagulants for treating HIT can be divided into: (a) long-acting, indirect (antithrombin-dependent) factor Xa inhibitors (danaparoid, fondaparinux) and (b) short-acting direct thrombin inhibitors (DTIs). (In theory, orally active direct factor Xa inhibitors, e.g. rivaroxaban, or DTIs, e.g. dabigatran, could be effective for treating HIT, but experience is not currently available.)
· Table 30.6 compares and contrasts the indirect factor Xa inhibitors versus the DTIs for the management of HIT and suspected HIT [8].
· The reader is referred elsewhere for dosing recommendations for the alternative nonheparin anticoagulants [18].
· HIT-associated consumptive coagulopathy can lead to treatment failure due to PTT-confounding [8,19].
· Fondaparinux is a reasonable option for treating HIT [20]; further, its proven efficacy and safety in numerous (non-HIT) indications of antithrombotic prophylaxis and therapy are important considerations, given that ∼90% of patients tested do not have HIT [19].
Table 30.6 A comparison of two classes of anticoagulant used to treat HIT.
|
Indirect (AT-dependent) factor Xa inhibitors: danaparoid, fondaparinux |
DTIs: argatroban, r-hirudin (lepirudin*, desirudin), bivalirudin |
|
|
Half-life |
✓ Long (danaparoid, 25 h†, fondaparinux, 17 h): reduces risk of rebound hypercoagulability |
Short (<2 h): potential for rebound hypercoagulability |
|
Dosing |
✓ Both prophylactic- and therapeutic-dose regimens‡ |
Prophylactic-dose regimens are not established (exception: subcutaneous desirudin) |
|
Monitoring |
✓ direct (antifactor Xa levels); accurate drug levels obtained |
Indirect (APTT): risk for DTI underdosing due to APTT elevation caused by non-DTI factors (‘APTT confounding’) |
|
Effect on INR |
✓ No significant effect; simplifies overlap with warfarin |
Increases INR: argatroban > bivalirudin > r-hirudin; complicates warfarin overlap |
|
Protein C pathway |
✓ No significant effect |
Thrombin inhibition could impair activation of protein C pathway |
|
Reversibility of action |
✓ Irreversible inhibition: AT forms covalent bond with factor Xa |
Irreversible inhibition only with r-hirudin |
|
Efficacy and safety established for non-HIT indications |
✓ Treatment and prophylaxis of VTE (danaparoid, fondaparinux) and ACS (fondaparinux) |
Not established for most non-HIT settings |
|
Platelet activation |
✓ Danaparoid inhibits platelet activation by HIT antibodies (fondaparinux has no effect) |
No effect |
|
Major bleeding risk |
✓ Relatively low |
Relatively high (∼1% per treatment day) |
|
Availability of antidote |
No |
No |
|
Inhibition of clot- bound thrombin |
No effect |
✓ Inhibits clot-bound thrombin |
|
Regulatory approval to treat HIT |
Danaparoid: yes (although not in the USA); fondaparinux: no |
Argatroban: yes. Lepirudin: yes. Bivalirudin: no. Desirudin: no. |
|
Drug clearance |
Predominantly renal |
Variable (predominantly hepatobiliary: argatroban; predominantly renal: r-hirudin) |
|
Check mark (✓) indicates favourable feature in comparison of drug classes. |
||
Prevention of warfarin-induced venous limb gangrene
· Warfarin and other vitamin K antagonists are contraindicated during the acute thrombocytopenic phase of HIT [18]. This is because their use is strongly associated with the risk of precipitating venous limb gangrene and (less often) central necrosis of skin and subcutaneous tissues (‘classic’ warfarin-induced skin necrosis) [13].
· Vitamin K should be given (5 to 10 mg by slow intravenous injection) if HIT is diagnosed in a patient who is receiving warfarin, especially if DTI therapy is planned (warfarin raises the APTT and thus risks APTT confounding of DTI therapy) [8,18,19].
· Prothrombin complex concentrates (PCCs) contain small amounts of heparin, and thus their use is relatively contraindicated during acute HIT.
· Argatroban–warfarin overlap is problematic because argatroban prolongs the INR. In a patient who bleeds while receiving argatroban, plasma or PCCs should not be given to reverse a very high INR because the coagulopathy is caused by argatroban rather than because of factor deficiency.
Management of isolated HIT
· ‘Isolated HIT’ is defined as HIT recognized because of thrombocytopenia, rather than because of a thrombotic event that draws attention to the possibility of HIT [11].
· Isolated HIT managed by simple discontinuation of heparin is associated with a ∼50% risk of symptomatic thrombosis (most often VTE) and 5% risk of sudden death due to pulmonary embolism [11]; thus, a rapidly acting alternative anticoagulant is recommended when isolated HIT is strongly suspected or confirmed.
· Our practice is to continue therapeutic-dose alternative anticoagulation until there is recovery of the platelet count to a stable plateau within the normal range; we then repeat the venous ultrasound and, if it is still negative for DVT, we discontinue anticoagulation.
Adjunctive therapies
· Thromboembolectomy sometimes can salvage an ischaemic limb due to acute large-vessel artery occlusion by platelet-rich ‘white clots’. Nonheparin anticoagulant protocols, however, are not well-established for vascular surgery.
· High-dose intravenous immunoglobulin (IVIgG) interferes with HIT antibody-induced platelet activation in vitro and reports indicate that its use can result in a platelet count increase in HIT. However, IVIgG is not an anticoagulant and its use should be considered adjunctive in special circumstances (e.g. severe, persisting HIT).
· Thrombolytic therapy may be considered in selected patients with limb- or organ-threatening thrombosis. Concomitant anticoagulation with a nonheparin anticoagulant should be administered if heparin is part of the standard thrombolysis protocol.
· Inferior vena cava filters should be avoided because their use contributes to local thrombus formation/extension and risks underutilization of anticoagulation, increasing chance of limb necrosis.
Repeat heparin exposure
· The immunology of HIT differs from the ‘classic’ immune response.
· Antibody titres decrease rapidly with cessation of HIT and in >60% of patients antibodies are no longer detectable after 100 days.
· In a patient with previous HIT who has become antibody-negative, re-exposure to heparin does not result in an anamnestic immune response. If repeat immunization occurs, at least 4–5 days are needed before antibodies are present in sufficient amounts to induce platelet activation.
· The low risk of triggering recurrent HIT allows for deliberate re-exposure to heparin for intraoperative anticoagulation during cardiac or vascular surgery [12,18]. Usually, heparin is avoided before and after surgery (if antibodies are regenerated, HIT is unlikely to be retriggered in the absence of further postoperative heparin use).
· Preliminary data from Belgium and Japan indicate that patients who develop HIT in association with chronic dialysis can be re-exposed to long-term intermittent heparin safely and without an anamnestic response, once PF4/heparin antibodies are no longer detectable [19].
Key points
1. HIT is a highly prothrombotic, antibody-mediated adverse effect of heparin.
2. Venous thrombosis occurs most often, especially DVT and pulmonary embolism; unusual venous thrombotic events include adrenal haemorrhagic necrosis (secondary to adrenal vein thrombosis) and cerebral venous (dural sinus thrombosis). Arterial thrombosis most often involves large limb arteries, cerebral arteries and coronary arteries.
3. The frequency of HIT varies widely and occurs more often in patients who receive UFH (versus LMWH) and are postoperative (versus medical, obstetric or pediatric); there is minor female predominance.
4. HIT is caused by IgG class antibodies that strongly activate platelets, triggering a procoagulant platelet response; almost always, the antibodies recognize multimolecular PF4/heparin complexes (the antibodies recognize one or more epitopes on PF4, as heparin can be substituted by certain other polyanions).
5. Washed platelet activation assays have the highest sensitivity-specificity trade-off for detecting HIT antibodies; although PF4-dependent ELISAs have high sensitivity for detecting HIT antibodies, they lack diagnostic specificity (except when strong positive ELISA results are observed, e.g. >2.00 optical density units in an IgG-specific ELISA).
6. HIT lacks features of a ‘classic’ immune response, i.e. antibodies of IgG class are detectable 4–5 days following an immunizing heparin exposure, without IgM precedence.
7. HIT antibodies are remarkably transient, accounting for why rapid-onset HIT only occurs in patients who have been exposed to heparin within the recent past. Also, it explains why heparin re-exposure is appropriate for patients with a previous history of HIT who require cardiac or vascular surgery, provided that platelet-activating antibodies are no longer detectable.
8. Vitamin K antagonists (e.g. warfarin) are contraindicated during the acute phase of HIT because their use can precipitate limb necrosis due to microthrombosis; vitamin K should be administered to a patient diagnosed with acute HIT who is receiving warfarin therapy.
9. Prophylactic platelet transfusions should be avoided during acute HIT, as thrombocytopenic bleeding (e.g. mucocutaneous hemorrhage) is not a feature of HIT and platelet transfusions in theory could increase thrombotic risk.
10. Treatment of HIT should focus on rapidly acting, nonheparin anticoagulants. There are two main classes of therapies: (a) long-acting indirect (antithrombin-dependent) factor Xa inhibitors (danaparoid, fondaparinux) and (b) direct thrombin inhibitors (argatroban, recombinant hirudin, bivalirudin).
References
1. Warkentin TE, Chong BH & Greinacher A. Heparin-induced thrombocytopenia: towards consensus. Thromb Haemost 1998; 79: 1–7.
2. Greinacher A, Kohlmann T, Strobel U, Sheppard JI & Warkentin TE. The temporal profile of the anti-PF4/heparin immune response. Blood 2009; 113: 4970–4976.
3. Warkentin TE, Sheppard JI, Moore JC, Cook RJ & Kelton JG. Studies of the immune response in heparin-induced thrombocytopenia. Blood 2009; 113: 4963–4969.
4. Krauel K, Pötschke C, Weber C, Kessler W, Fürll B, Ittermann T et al. Platelet factor 4 binds to bacteria, inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood 2011; 117: 1370–1378.
5. Warkentin TE & Kelton JG. Delayed-onset heparin-induced thrombocytopenia and thrombosis. Ann Int Med 2001; 135: 502–506.
6. Warkentin TE, Sheppard JI, Sigouin CS, Kohlmann T, Eichler P & Greinacher A. Gender imbalance and risk factor interactions in heparin-induced thrombocytopenia. Blood 2006; 108: 2937–2941.
7. Warkentin TE, Makris M, Jay RM & Kelton JG. A spontaneous prothrombotic disorder resembling heparin-induced thrombocytopenia. Am J Med 2008; 121: 632–636.
8. Warkentin TE. Agents for the treatment of heparin-induced thrombocytopenia. Hematol/Oncol Clin N Am 2010; 24: 755–775.
9. Warkentin TE & Linkins LA. Immunoassays are not created equal. J Thromb Haemost 2009; 7: 1256–1259.
10. Bakchoul T, Giptner A, Bein G, Santoso S & Sachs UJH. Prospective evaluation of immunoassays for the diagnosis of heparin-induced thrombocytopenia. J Thromb Haemost 2009; 7: 1260–1265.
11. Warkentin TE & Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med 1996; 101: 502–507.
12. Warkentin TE & Kelton JG. Temporal aspects of heparin-induced thrombocytopenia. N Engl J Med 2001; 344: 1286–1292.
13. Warkentin TE, Elavathil LJ, Hayward CPM, Johnston MA, Russett JI & Kelton JG. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann Int Med 1997; 127: 804–812.
14. Warkentin TE & Linkins LA. Non-necrotizing heparin-induced skin lesions and the 4T's score. J Thromb Haemost 2010; 8: 1483–1485.
15. Selleng S, Malowsky B, Strobel U, Wessel A, Ittermann T, Wollert HG et al. Early-onset and persisting thrombocytopenia in post-cardiac surgery patients is rarely due to heparin-induced thrombocytopenia even when antibody tests are positive. J Thromb Haemost 2010; 8: 30–36.
16. Warkentin TE, Sheppard JI, Moore JC, Sigouin CS & Kelton JG. Quantitative interpretation of optical density measurements using PF4-dependent enzyme-immunoassays. J Thromb Haemost 2008; 6: 1304–1312.
17. Nellen V, Sulzer I, Barizzi G, Lämmle B & Alberio L. Rapid exclusion or confirmation of heparin-induced thrombocytopenia: a single-centre experience with 1291 patients. Haematologica 2012; 97: 89–97.
18. Warkentin TE, Greinacher A, Koster A & Lincoff AM. Treatment and prevention of heparin-induced thrombocytopenia. American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008; 133(6 Suppl.): 340S–80S.
19. Warkentin TE. HIT paradigms and paradoxes. J Thromb Haemost 2011; 9(Suppl. 1): 105–117.
20. Warkentin TE, Pai M, Sheppard JI, Schulman S, Spyropoulos AC & Eikelboom JW. Fondaparinux treatment of acute heparin-induced thrombocytopenia confirmed by the serotonin-release assay: a 30-month, 16-patient case series. J Thromb Haemost 2011; 9: 2389–2396.
Further reading
Arepally GM & Ortel TL. Clinical practice. Heparin-induced thrombocytopenia. N Engl J Med 2006; 355: 809–817.
Cuker A & Cines DB. How I treat heparin-induced thrombocytopenia. Blood 2012; 119: 2209–2218.
Greinacher A. Heparin-induced thrombocytopenia. J Thromb Haemost 2009; 7(Suppl. 1): 9–12.
Greinacher A, Pötzsch B, Amiral J, Dummel V, Eichner A & Mueller-Eckhardt C. Heparin-associated thrombocytopenia: isolation of the antibody and characterization of a multimolecular PF4-heparin complex as the major antigen. J Thromb Haemost 1994; 71: 247–251.
Greinacher A, Holtfreter B, Krauel K, Gätke D, Weber C, Ittermann T et al. Association of natural anti-platelet factor 4/heparin antibodies with periodontal disease. Blood 2011; 118: 1395–1401.
Lubenow N, Hinz P, Thomaschewski S, Lietz T, Vogler M, Ladwig A et al. The severity of trauma determines the immune response to PF4/heparin and the frequency of heparin-induced thrombocytopenia. Blood 2010; 115: 1797–1803.
Warkentin TE & Greinacher A (eds). Heparin-Induced Thrombocytopenia, 5th edn. Boca Raton, Florida: CRC Press, 2013 (641 pages).
Warkentin TE & Greinacher A. Heparin-induced anaphylactic and anaphylactoid reactions: two distinct but overlapping syndromes. Expert Opin Drug Saf 2009; 8: 129–144.
Warkentin TE & Sheppard JI. Testing for heparin-induced thrombocytopenia antibodies. Transfus Med Rev 2006; 20: 259–272.
Warkentin TE, Cook RJ, Marder VJ, Sheppard JI, Moore JC, Eriksson BI et al. Anti-platelet factor 4/heparin antibodies in orthopedic surgery patients receiving antithrombotic prophylaxis with fondaparinux or enoxaparin. Blood 2005; 106: 3791–3796.