Siraj A. Misbah
Oxford University Hospitals, University of Oxford, Oxford, UK
Introduction
The increasing awareness of immunodeficiency and the rapid pace of genetic discovery have helped to ensure that immunodeficiency disorders are no longer viewed as arcane rarities by both clinical immunologists and nonimmunologists. In haematology, alongside the major changes in practice that have been driven by advances in fundamental immunology [1], haematologists are also likely to encounter patients with primary immunodeficiency disease because of the frequency of haematological complications associated with this group of disorders. Given that most haematologists will be familiar with the consequences of secondary immunodeficiency, either iatrogenic or associated with lymphoproliferative disease, this chapter will focus primarily on primary immunodeficiency disorders followed by a separate section on immunoglobulin therapy.
Primary immunodeficiency disorders
Many primary immunodeficiency disorders associated with single gene mutations have been aptly called experiments of nature in view of the unique insights that these diseases have provided in unravelling complex immunological functions. Currently, the World Health Organization – International Union of Immunological Societies (WHO/IUIS) committee on primary immunodeficiency diseases recognizes over 150 primary immunodeficiencies for which the underlying molecular basis has been elucidated [2]. As the genetic basis of old and new immunodeficiency disorders is unravelled, it has become clear that the same gene mutation may result in different phenotypes. In investigating and managing patients with primary immunodeficiencies, it is important to bear in mind this concept of genetic heterogeneity accompanied by equally significant clinical and immunological heterogeneity. For example, the same mutation in the gene encoding the Wiskott–Aldrich syndrome protein (WASP) may result in either full-blown Wiskott–Aldrich syndrome characterized by thrombocytopenia, infections and autoimmunity or a limted phenotype of X-linked thrombocytopenia [3]. Such examples have focused attention on the role of epigenetic changes in influencing disease phenotype.
Although primary immunodeficiencies can affect any part of the immune system, in practice patients with predominant defects of B-cell function and combined B- and T-cell defects constitute the bulk of a clinical immunologist's workload. The immunopathogenesis of antibody deficiency disorders and combined B- and T-lymphocyte deficiency is best understood within the context of B- and T-lymphocyte development. Whilst a detailed discussion of B- and T-cell development is outside the scope of this chapter, the schematic diagrams set out in Figures 31.1 and 31.2 summarize the major events in B- and T-cell development and the points at which developmental arrest leads to immunodeficiency.
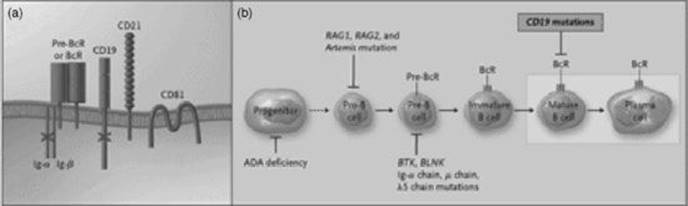
Fig 31.1 Mutations in multiple proteins, including the CD3 and ζ chains, that cause T-cell immunodeficiencies. The pre-T-cell receptor (pre-TcR) and mature TcR complexes consist of a receptor dimer associated with CD3 chains γ, δ and ɛ and a ζ-chain dimer (panel (a)). The pre-TcR complex differs from the mature complex owing to the presence of a surrogate chain (indicated by a dotted line) in the pre-TcR dimer. The CD3 and ζ chains facilitate the expression of the complex on the cell surface and send intracellular signals. Mutations in the receptor complexes that have been linked to T-cell immunodeficiencies are indicated with a bold X. T-cell differentiation (panel (b)) entails the progression from a progenitor cell to a CD4–CD8– thymocyte that expresses a pre-TcR, followed by differentiation into a CD4+CD8+ thymocyte expressing the mature TcR. This cell develops into a single CD4+CD– or CD4–CD+ thymocyte and, finally, into a CD4+CD8– or CD4–CD8+ mature T cell. Dashed lines indicate that intervening steps occur that are not shown. The stages of differentiation affected by mutations and deficiencies of different proteins are shown by T bars. Bold bars indicate a partial effect. ADA, adenosine deaminase; JAK3, Janus kinase 3; RAG, recombination-activating gene; and ZAP-70, zeta-chain-associated protein of 70 kDa. Reproduced with permission from Rudd CE. N Engl J Med 2006; 354: 1874. Copyright 2006 Massachusetts Medical Society. All rights reserved.
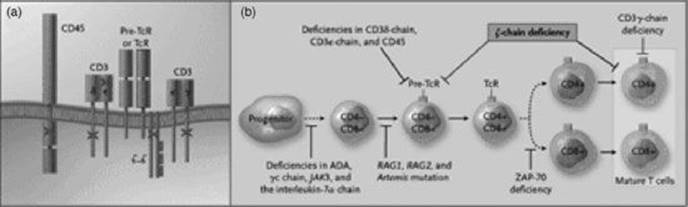
Fig 31.2 Mutations in multiple proteins, including pre-B-cell receptor (pre-BcR) and CD19, that cause B-cell immunodeficiencies. The pre-BcR and mature BcR complexes consist of an immunoglobulin dimer associated with the lg-α and lg-β subunits that generate intracellular signals (panel (a)). The pre-BcR differs from the mature complex owing to the presence of a surrogate light chain (indicated by a dotted line) in the pre-BcR dimer. Further associated with the BcR are CD19, CD21, CD81 (TAPA-1) and CD225 (Leu-13, not shown), which act as coreceptors to modulate the threshold of signalling. Mutations in the receptor complexes that have been linked to B-cell immunodeficiencies are indicated with a bold X. B-cell differentiation (panel (b)) entails a progression from a progenitor stem cell to a pro-B cell to a pre-B cell to an immature B cell and, finally, to a mature B cell. The dashed line indicates that intervening steps occur that are not shown. The pre-BcR provides signals for pre-B-cell differentiation. The stages of B-cell differentiation affected by mutations and deficiencies of different proteins are shown by T bars. ADA, adenosine deaminase; RAG, recombination-activating gene; BTK, Bruton's tyrosine kinase; and BLNK, mutated B-cell-linked protein. Reproduced with permission from Rudd CE. N Engl J Med 2006; 354: 1875. Copyright 2006 Massachusetts Medical Society. All rights reserved.
Predominant B-cell deficiency disorders
Common variable immunodeficiency
Of the 20 antibody deficiency disorders currently recognized, common variable immunodeficiency (CVID) is the commonest acquired primary immunodeficiency that is likely to be encountered by haematologists. As its name implies, CVID is characterized by a severe reduction in at least two serum immunoglobulin isotypes associated with low or normal B-cell numbers. In contrast, antibody deficiency disorders associated with severe reduction of all serum immunoglobulin isotypes with absent circulating B cells is a feature of diseases associated with mutations that interrupt B-cell development (Figure 31.2).
The term CVID embraces a heterogeneous group of disorders, all of which are characterized by late-onset hypogammaglobulinaemia as the unifying theme [4]. The commonest infective manifestation of antibody deficiency is recurrent infection with encapsulated bacteria, particularly Streptococcus pneumoniae and to a lesser extent with unencapsulated Haemophilus influenzae. Many patients develop frank bronchiectasis as a consequence of recurrent chest infections. Despite their inability to mount effective antibody responses to exogenous pathogens many patients with CVID mount paradoxical immune responses to self-antigens leading to autoimmune disease. In a haematological context, the most frequent of these autoimmune complications are immune thrombocytopenic purpura (ITP) and autoimmune haemolytic anaemia.
A whole host of other organ-specific and systemic autoimmune diseases may also occur, ranging from Addison's disease to systemic lupus erythematosus. Other noninfective complications associated with CVID include a curious predisposition to granulomatous disease, lymphoid interstitial pneumonitis and a 100-fold increase in the risk of lymphoma. Although the latter may occasionally be driven by Epstein–Barr virus (EBV), in the majority of cases no underlying infection is evident, raising the possibility that lymphoproliferative disease in these patients is a manifestation of defective immunoregulation.
Despite the inability of B cells in CVID to produce antibodies, recovery of antibody production has been documented following infection with HCV and HIV, respectively [5]. This observation supports the concept that defective immunoregulation is contributing to poor B-cell function in these patients.
Given the range of infective and noninfective complications associated with CVID, many attempts have been made to produce a clinically useful disease classification based on immunological indices. Recent evidence suggests that a deficiency of switched IgM− IgD− CD27+ memory B cells may well correlate with the development of bronchiectasis, autoimmunity and reactive splenomegaly in CVID. The molecular basis for some of the diseases previously included under the umbrella of CVID has recently been elucidated by the detection of mutations in a number of genes associated with B-cell function (Table 31.1). In addition to the molecular defects listed in Table 31.1, there are rare patients with mutations in certain X-linked genes (Bruton tyrosine kinase, CD40 ligand and signalling lymphocyte activation – associated protein) who may present with a clinical phenotype resembling CVID.
Table 31.1 Known molecular defects that present with a CVID-like clinical picture.
|
· Inducible costimulatory receptor (ICOS) deficiency · CD19 deficiency · Mutations in the transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI) receptor · Mutations in the receptor for B-cell activating factor of the TNF family (BAFF) |
The management of CVID revolves around regular immunoglobulin replacement optimized to ensure a trough IgG level well within the normal range for effective prophylaxis against bacterial infections. Evidence from a longitudinal study of infection outcomes in 90 patients with CVID followed up over 20 years suggests that the dose of immunoglobulin required to reduce breakthrough infections is individual to a particular patient [6]. Achievement of this goal, therefore, is likely to be associated with a wide range of trough IgG levels. Early diagnosis and therapeutic intervention with immunoglobulin therapy significantly minimizes the risk of permanent bronchiectatic lung damage.
X-linked agammaglobulinaemia
X-linked agammaglobulinaemia (XLA) was one of the earliest primary immunodeficiencies to be clinically characterized in the 1950s. Its molecular basis was only elucidated in the 1990s with the discovery of mutations in a protein tyrosine kinase gene, named Bruton's tyrosine kinase (Btk).
The Btk gene is located on the long arm of the X-chromosome and encodes for a cytoplasmic tyrosine kinase, which is essential for B-cell signal transduction. Btk mutations are associated with B-cell developmental arrest in the bone marrow. The consequent disappearance of circulating B cells in association with severe panhypogammaglobulinaemia and poorly developed lymphoid tissue constitutes the cardinal immunological features of XLA. Over 400 different mutations in the Btk gene have been recorded to date but there are no significant correlations between genotype and clinical phenotype. The essential role of Btk in B-cell receptor signal transduction, as exemplified by B-cell failure in XLA, is currently being exploited by the development of Btk inhibitors for the treatment of B-cell lymphomas.
Most boys with XLA present with a history of recurrent sinopulmonary infections on a background of panhypogammaglobulinaemia after the age of 6 months, once the protective effect of transplacentally acquired maternal IgG has waned. As with CVID, delayed diagnosis of XLA and consequent failure to institute adequate immunoglobulin replacement is associated with a high risk of bronchiectasis [7].
In keeping with the absence of a T-cell defect in XLA, infection with intracellular pathogens is generally not a problem. The major exception to this rule is the predisposition to chronic enteroviral infections, including echovirus meningoencephalitis and vaccine-induced poliomyelitis. A clinical phenotype identical to XLA may be caused by mutations in the μ-immunoglobulin heavy-chain gene and other components of the B-cell receptor [8].
Severe combined immunodeficiency
Severe combined immunodeficiency (SCID) refers to a group of genetically determined disorders characterized by arrested T-cell development accompanied by impaired B-cell function [9]. The incidence of SCID is estimated to be between 1:50 000 and 1:100 000 live births.
Babies with SCID present with recurrent infections associated with lymphopenia. Among the range of pathogens responsible for infection in SCID, Pneumocystis jiroveci (carinii), aspergillus species and cytomegalovirus predominate in keeping with the profound T-cell deficiency seen in these babies.
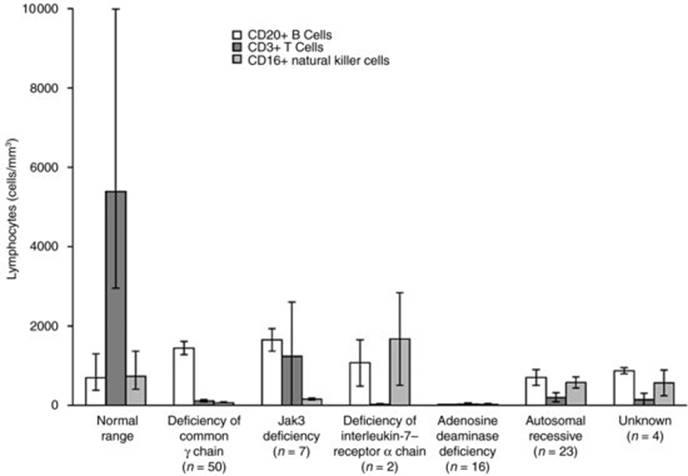
To date, at least 11 distinct molecular defects that cause the SCID phenotype have been identified (Table 31.2). Whilst lymphopenia is characteristic of all forms of SCID (Figure 31.3), the circulating lymphocyte surface marker profile (Table 31.2) provides a useful clue as to the underlying genetic defect. For example, deficiency of adenosine deaminase, a key purine enzyme, results in severe lymphopenia affecting T, B and NK cells leading to its characterization as T–B–NK–SCID.
Table 31.2 Classification of severe combined immunodeficiency.
|
Affected gene |
Inheritance |
Circulating lymphocyte phenotype |
|
Adenosine deaminase (ADA) |
AR |
T– B– NK– |
|
Common cytokine γ-chain (γc) |
X-linked |
T– B+ NK– |
|
Jak-3 |
AR |
T– B+ NK– |
|
IL-7α |
AR |
T– B+ NK+ |
|
Recombination activating gene 1,2 (RAG1/RAG2) |
AR |
T– B– NK+ |
|
Artemis |
AR |
T– B– NK+ |
|
CD3 δ, ζ, ɛ |
AR |
T– B+ NK+ |
|
CD45 |
AR |
T– B+ NK+ |
|
AR, autosomal recessive. |
||
Fig 31.3 Mean (± SE) numbers of CD20+ B cells, CD3+ T cells and CD16+ natural killer cells at presentation in 102 patients with severe combined immunodeficiency, according to the cause of the disorder. The lymphopenia characteristic of all forms of severe combined immunodeficiency is apparent, as are the differences in the lymphocyte phenotypes in the various forms of the syndrome. The normal ranges at the author's institution are shown for comparison. Jak3 denotes Janus kinase 3. ‘Autosomal recessive’ refers to 23 patients with autosomal recessive severe combined immunodeficiency in whom the molecular defect has not been identified. Reproduced with permission from Buckley RH. N Engl J Med 2000; 343: 1314. Copyright 2000 Massachusetts Medical Society. All rights reserved.
Given the profound impairment in T-cell immunity, babies with SCID are at risk of iatrogenic disease with live vaccines and transfusion-associated graft-versus-host disease. For these reasons, immunization with live vaccines should be regarded as absolutely contraindicated in these babies. Equally, any baby with SCID should only receive irradiated and cytomegalovirus-seronegative blood.
The severity of disease and the urgency with which curative haemopoietic stem cell transplantation (HSCT) should be undertaken has led SCID to be regarded as a paediatric emergency. The results of HSCT have improved significantly with early diagnosis and aggressive management of infections and nutritional problems seen in these babies at the time of diagnosis. At present, HSCT from an HLA-matched sibling donor offers an 80% chance of cure whilst a fully HLA-matched unrelated transplant offers a 70% chance of cure (Figure 31.4). Neonatal screening for SCID using polymerase chain reaction-based analysis of T-cell receptor excision circles (TRECs – a measure of thymic Tcell output) on Guthrie card blood samples has recently been introduced in parts of the USA [10].
Fig 31.4 Cumulative probability of survival in SCID patients, according to donor source (related or unrelated donor) and HLA matching, and year of transplantation. Reproduced with permission from Antoine C et al. Lancet 2003; 361: 556.

In view of the single gene defects underlying SCID, gene therapy is an attractive option. Whilst offering great promise, the results of gene therapy to date have been mixed. Gene therapy has been successful in some children with ADA and common cytokine γ-chain deficiency, respectively, with evidence of T-, B- and NK-cell reconstitution in the former and T- and NK-cell reconstitution in the latter. However, the occurrence of insertional mutagenesis leading to T-cell lymphoproliferative disease in some children with common γ-chain SCID is an important reminder of the obstacles associated with this ground-breaking therapy [11].
Investigation of suspected immunodeficiency
Although a few patients may have distinctive clues on examination pointing towards an immunodeficiency, most patients have no physical signs that would specifically point to an immunodeficiency disorder. Conversely, it follows that a normal physical examination does not exclude immunodeficiency disease.
Immunodeficiency should be included in the differential diagnosis of any patient with severe, prolonged or recurrent infection with common pathogens or even a single episode of infection with an unusual pathogen. The type of pathogen involved provides important clues as to which component of the immune system may be defective and consequently guides the selection of relevant immunological tests (Table 31.3). Although this chapter is primarily devoted to primary immunodeficiency, it is essential to consider and exclude the possibility of HIV infection as a driver for immunodeficiency in many of these clinical scenarios [12].
Table 31.3 Patterns of infection as a guide to selection of immunological tests in suspected immune deficiency.
|
Type of pathogen |
Consider |
Relevant immunological tests |
|
A – Encapsulated pathogens |
Antibody deficiency Complement deficiency |
Serum immunoglobulins, specific antibodies to polysaccharide and protein antigens |
|
Haemolytic complement activity |
||
|
B – Viruses and intracellular pathogens |
T-cell defect |
Lymphocyte surface marker analysis |
|
Lymphocyte transformation |
||
|
C – Combination of encapsulated pathogens and viruses and other intracellular pathogens |
Combined B- + T-cell defect |
As for A and B |
|
D – Recurrent neisserial infection |
Complement deficiency |
Haemolytic complement activity |
|
E – Recurrent staphylococcal abscesses and/or invasive fungal infections |
Phagocyte defect |
Neutrophil respiratory burst |
|
Leucocyte adhesion molecule expression (selected cases) |
In view of the complexity of many immunological tests it is essential that immunological investigations are performed under the guidance of a clinical immunologist to enable appropriate test selection, interpretation and advice on clinical management.
Management of immunodeficiency
Infections in any immunodeficient patient should be treated aggressively with appropriate antimicrobial therapy. In patients with antibody deficiency, lifelong immunoglobulin replacement remains the cornerstone of management. For children with SCID, HSCT remains the main curative option with the prospect of gene therapy for some forms of SCID. Patients with complement deficiency should be fully immunized with the full range of available vaccines against neisserial, pneumococcal and haemophilus infections. However, it is vital to avoid the use of live vaccines in any patient with immunodeficiency in view of the real risks of vaccine-associated disease, as exemplified by vaccine-induced poliomyelitis in XLA and BCG-induced mycobacterial disease in SCID.
Immunoglobulin therapy
Therapeutic immunoglobulin is a blood component prepared from the plasma of 10 000–15 000 donors. The broad spectrum of antibody specificities contained in pooled plasma is an essential ingredient underpinning the success of intravenous (IVIg) and, more recently, subcutaneous immunoglobulin in infection prophylaxis in patients with antibody deficiency. Evidence from longitudinal studies in large cohorts of antibody-deficient patients and a meta-analysis of studies of IVIg replacement have highlighted the inverse correlation between trough IgG levels and the frequency of infection [13]. A similar inverse relationship between incidence of infections and steady-state IgG levels has recently been confirmed in studies of subcutaneous immunoglobulin in patients with primary immunodeficiency. In addition to its role in straightforward antibody replacement, the success of high dose IVIg in the treatment of ITP has led to a veritable explosion in its use as a therapeutic immunomodulator in many autoimmune diseases spanning multiple specialties (Table 31.4).
Table 31.4 Use of IVIg as an immunomodulatory agent.
|
Disorder |
Comments |
|
Neurology |
|
|
Guillain–Barre syndrome |
Treatment of choice and as efficacious as plasmapheresis (RCT, CR) |
|
Multifocal motor neuropathy |
Treatment of choice (RCT) |
|
Chronic inflammatory demyelinating polyneuropathy |
As an alternative to steroids (RCT) |
|
Dermatomyositis |
As an adjunct to immunosuppressive therapy (RCT) |
|
Myasthenia gravis |
For myasthenic crises (RCT) |
|
Lambert–Eaton syndrome |
For non-cancer-associated cases that have failed to respond to standard therapy (RCT) |
|
Stiff-person syndrome |
For severe cases unresponsive to standard therapy (RCT) |
|
Haematology |
|
|
Immune thrombocytopenic purpura |
Selected cases unresponsive to standard treatment (RCT) |
|
Parvovirus-associated pure red cell aplasia |
Selected cases |
|
Paediatrics |
|
|
Kawasaki disease |
Treatment of choice (RCT) |
|
Dermatology |
|
|
Toxic epidermal necrolysis |
Open studies/case series suggest benefit |
|
Autoimmune blistering disorders |
Open studies/case series suggest benefit |
|
Streptococcal toxic shock syndrome |
Open studies/case series suggest benefit |
|
The list of indications is not exhaustive but covers those disorders where IVIg is frequently used. RCT – evidence from randomized controlled trials; CR – evidence from Cochrane review. |
|
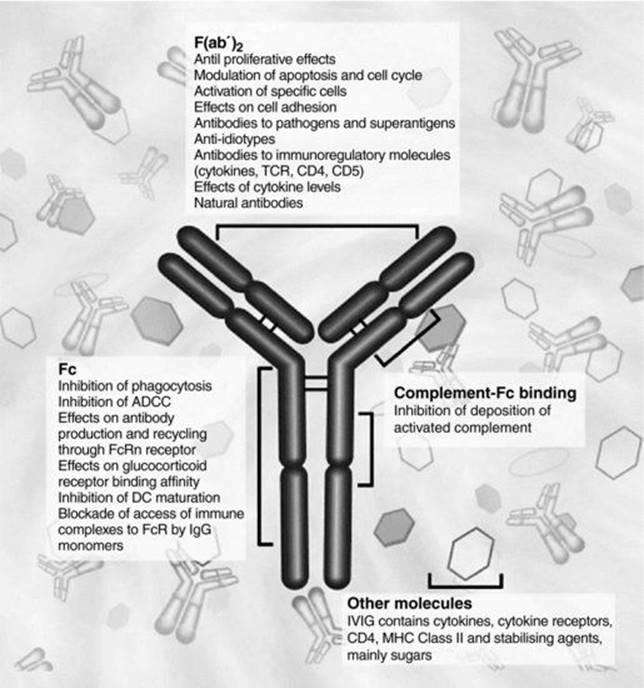
The mechanisms of action of high dose IVIg in autoimmune disease are complex and reflect the potent immunological actions of the different regions of an IgG molecule. It is helpful conceptually to consider the potential mechanisms of action in relation to the variable regions of IgG (F(ab′)2), the Fc region and the presence in IVIg of other potent immunomodulatory substances other than antibody (Figure 31.5). In ITP, the traditional view of Fc-receptor blockade as the predominant mechanism by which IVIg is effective has recently been complemented by evidence from murine studies showing that IVIg-mediated amelioration of ITP is crucially dependent on interactions with the inhibitory FcγRIIB as well as the activating receptor, FcγRIII. Evidence from murine studies indicates that upregulation of FcγRIIB expression occurs via the small sialylated immunoglobulin component of polyclonal IVIg (estimated at 1–3%). This observation has led to the hypothesis that the use of small doses of concentrated sialylated immunoglobulin might be as efficacious as the use of conventional high dose immunoglobulin for immunomodulation [14].
Fig 31.5 Immunomodulatory actions of intravenous immunoglobulin. Reproduced with permission from Jolles S, Sewell WAC & Misbah SA. Clin Exp Immunol 2005; 142: 3.
Immunoglobulin replacement in secondary antibody deficiency
IVIg replacement is beneficial in prophylaxis against infection in selected patients with secondary antibody deficiency associated with B-cell lymphoproliferative disease and myeloma. The predictors of response to IVIg are the presence of hypogammaglobulinaemia accompanied by low concentrations of pneumococcal antibodies and a failure to respond to test immunization with pneumococcal polysaccharide (Pneumovax). Whilst IVIg is clinically efficacious in patients fulfilling the above criteria, questions remain regarding its overall cost effectiveness [15]. For this reason, IVIg replacement should be reserved for those patients who have failed a trial of prolonged antibiotic prophylaxis. Despite evidence supporting the use of IVIg in secondary antibody deficiency, in practice its use has not been widespread due to the advent of more immunogenic pneumococcal conjugate vaccines coupled with improved overall management of these haematological malignancies.
Adverse effects of intravenous immunoglobulin therapy
Immediate infusion-related adverse effects
Minor to moderate immediate infusion-related adverse effects in the form of headaches, chills, rigors and backache occur in approximately 1% of patients irrespective of the therapeutic dose of immunoglobulin. These adverse effects are largely related to the rate of infusion and/or the presence of underlying infection in the recipient and respond to a combination of a reduction in infusion rate coupled with simple analgesia. Very rarely, some patients with total IgA deficiency and pre-existing anti-IgA antibodies may develop anaphylaxis on exposure to IVIg preparations containing IgA. This risk is greatly minimized by the use of an IgA-depleted IVIg preparation in such patients.
Dose-related adverse effects
The increasing use of IVIg for therapeutic immuno- modulation has been associated with the development of a range of haematological, neurological, nephrological and dermatological adverse effects that are directly linked to the high doses (2 g/kg) required for autoimmune disease in contrast to the low doses (0.4 g/kg) used for antibody replacement.
Haematological
High dose IVIg causes a dose-dependent increase in plasma viscosity [16], which is sufficient to precipitate serious arterial and venous thrombosis in patients with pre-existing thrombophilia, paraproteinaemia, severe polyclonal hypergammaglobulinaemia and atheromatous cardiovascular disease.
The risk of IVIg-associated acute haemolysis due to passive transmission of anti-blood-group antibodies has been greatly minimized by the institution of rigorous quality control measures designed to ensure that the titre of anti-blood-group antibodies in IVIg does not exceed 1:8.
Neurological
High dose IVIg is associated with the development of self-limiting acute aseptic meningitis in a minority of patients (<5%). Patients with background migraine are at higher risk, raising the possibility that meningeal irritation may be due to the interaction of exogenous IgG with meningeal endothelium.
Renal
Nephrotoxicity due to high dose IVIg is a particular risk associated with sucrose-containing preparations, which trigger osmotic tubular injury leading to extensive vacuolar changes suggestive of historical cases of sucrose-induced nephropathy. The risk of renal damage is greatly minimized by avoiding the use of sucrose-containing IVIg preparations in patients with pre-existing diabetes and renal disease.
IVIg should also be avoided or used with caution in patients with mixed cryoglobulinaemia because of the real risk of the IgM component of cryoglobulin, containing rheumatoid factor reactivity complexing with infused exogenous IgG to cause acute immune-complex-mediated renal injury [17].
Dermatological
A variety of cutaneous adverse effects including eczema, erythema multiforme, urticaria and cutaneous vasculitis may be triggered by high dose IVIg. The relatively small number of cases reported to date does not enable any useful analysis that might help in minimizing the development of dermatological adverse reactions.
Risks of viral transmission
Viral transmission is a risk with both low and high dose IVIg therapy. However, the increasingly stringent screening of donors coupled with the introduction of additional antiviral steps during plasma fractionation has greatly reduced but not eliminated the risk of HCV transmission with IVIg. For this reason, patients on maintenance IVIg should have their liver function monitored along with regular testing for HCV. The lack of any outbreaks of IVIg-associated HCV transmission since the last outbreak in 1993 [18] attests to the success of current viral safety measures. Unlike HCV, HIV and HBV have never been transmitted by IVIg since the process of Cohn-ethanol fractionation specifically inactivates both of these viruses.
Whilst recent reports of the development of new variant Creutzfeldt–Jakob disease in recipients of blood from donors with asymptomatic disease have raised concerns of the possibility of prion transmission by blood components, this risk remains largely theoretical with IVIg. Leucocyte reduction and the use of plasma from countries free of bovine spongiform encephalopathy are measures designed to minimize this risk in the UK (see Chapter 15).
Practical aspects of immunoglobulin therapy – product selection and safe use
The availability of several different preparations of IVIg (at least six in the UK at present) has raised the question of whether IVIg should be considered to be a generic product. For the purposes of antibody replacement, it is reasonable to consider the different products equally efficacious since each product is required to fulfil the stringent criteria laid down by the World Health Organization for therapeutic immunoglobulin. With regard to the use of high dose IVIg as an immunomodulator, studies comparing the efficacy of different products in Kawasaki disease and chronic inflammatory demyelinating polyneuropathy (CIDP) have shown no difference in efficacy. Nonetheless, because differences in the manufacturing process affect opsonic activity, Fc-receptor function and complement fixation, it is best not to consider IVIg as a generic product. In view of this and the potential difficulty in tracking any future outbreak of IVIg-associated viral transmission, it is prudent to maintain patients requiring long-term treatment on the same IVIg product, irrespective of whether IVIg is being used for antibody deficiency or immunomodulation.
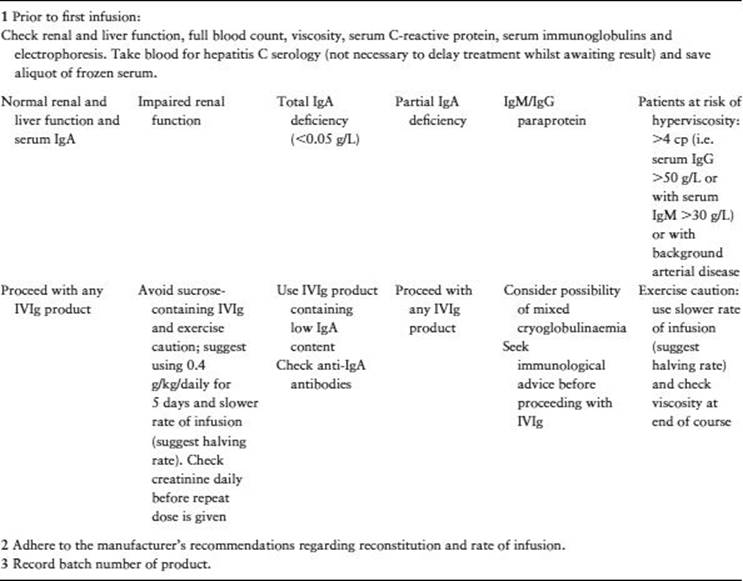
Table 31.5 provides a useful checklist for the safe use of high dose IVIg, including advice on product selection. Advice on individual products should be sought from a clinical immunologist.
Table 31.5 Checklist for the use of high dose IVIg. Reproduced with permission from Association of British Neurologists. Guidelines on IVIg in neurological diseases. http://www.theabn.org/.
Subcutaneous immunoglobulin
Following comparative trials, the subcutaneous route of immunoglobulin delivery has been shown to be as efficacious as IVIg in infection prophylaxis in patients with primary antibody deficiency [19]. In practice, SCIg has proven to be popular with both patients and clinicians in view of its ease of use in patients with poor venous access and minimal adverse effects, in comparison with IVIg (Table 31.6). Using a weekly infusion regimen, SCIg achieves steady-state IgG levels without the peaks and troughs associated with IVIg. When patients transfer from IVIg to SCIg, the achievement of equivalent or higher steady-state levels with the same dose of SCIg reflects the reduced catabolism with subcutaneous delivery.
Table 31.6 Adverse effects of intravenous versus subcutaneous immunoglobulin.
|
SCIg |
IVIg |
|
|
Local reactions at site of infusion |
Common (trivial) |
Nil |
|
Anaphylaxis |
— |
Very rare* |
|
Viral transmission (HCV) |
— |
+† |
|
Renal impairment |
— |
+ |
|
Aseptic meningitis |
— |
+ |
|
Thrombosis |
— |
+ |
|
Possibly related to anti-IgA abs in some cases. |
||
The success of SCIg as replacement therapy in antibody deficiency has led to its increasing use for immunomodulation as in multifocal motor neuropathy [20]. The use of multiple infusion sites in a motivated patient has enabled the delivery of higher doses required for immunomodulation. Using currently available 16% SCIg preparations, patients with autoimmune neuropathies are able to self-treat themselves with volumes of 200 to 220 mL weekly (32–35.2 g). The recent licensing of a 20% SCIg preparation and the future development of hyaluronidase-based preparations will enable the delivery of even higher doses and drive expansion of the immunomodulatory use of SCIg.
Key points
1. Over 150 primary immunodeficiency disorders are currently recognized.
2. Common variable immunodeficiency is the commonest acquired treatable immunodeficiency.
3. IVIg or SCIg is the mainstay of treatment for patients with antibody deficiency.
4. Haemopoietic stem cell transplantation remains the main curative option for children with SCID.
5. High dose IVIg is widely used as a therapeutic immunomodulator in a range of autoimmune diseases.
References
1. Caligaris-Cappio F. How immunology is reshaping clinical disciplines: the example of haematology. Lancet 2001; 358: 49–55.
2. Herz-Al W, Bousfiha A, Casanova JL et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Frontiers in Immunology 2011; 2: 1–26.
3. Buchbinder D, Nadeau K & Nugent D. Monozygotic twin pair showing discordant phenotype for X-linked thrombocytopenia and Wiskott–Aldrich syndrome: a role for epigenetics? J Clin Immunol 2011; 31: 773–777.
4. Cunningham-Rundles C. How I treat common variable immune deficiency. Blood 2010; 116: 7–15.
5. Jolles S, Tyrer M, Johnston M & Webster D. Long term recovery of IgG and IgM production during HIV infection in common variable immunodeficiency. J Clin Path 2001; 54: 713–715.
6. Lucas M, Lee M, Lortan J et al. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol 2010; 125: 1354–1360.
7. Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, Conley ME, Cunningham-Rundles C & Ochs HD. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore) 2006; 85: 193–202.
8. Ferrari S, Zuntini R, Lougaris V et al. Molecular analysis of the pre-BCR complex in a large cohort of patients affected by autosomal recessive agammaglobulinaemia. Genes Immunity 2007; 8: 325–333.
9. van der Burg M & Gennery AR. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur J Paed 2011; 170: 561–571.
10. Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T cell receptor excision circles. J Allergy Clin Immunol 2012; 129: 607–616.
11. Howe SJ, Mansour MR, Schwarzwaelder K et al. Insertional mutagenesis with acquired somatic mutations causes leukaemogenesis following gene therapy of SCID-X1 patients. J Clin Invest 2008; 118: 3143–3150.
12. Hanson IC & Shearer WT. Ruling out HIV infection when testing for severe combined immunodeficiencies and other T cell defects. J Allergy Clin Immunol 2012; 129: 875–876.
13. Orange JS, Grossman WJ, Navickis RJ et al. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol 2010;137: 21–30.
14. Anthony RM & Ravetch JV. A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol 2010; 30: S9–S14.
15. Raanani P, Gaffer-Gvili A, Paul M et al. Immunoglobulin prophylaxis in chronic lymphocytic leukaemia and multiple myeloma: systematic review and meta-analysis. Leuk Lymphoma 2009; 50: 764–772.
16. Bentley P, Rosso M, Sadnicka A et al. Intravenous immunoglobulin increases plasma viscosity without parallel rise in blood pressure. J Clin Pharm Ther 2011, 18 July. DOI: 10.1111/j.1365-2710.2011.01287.x. Epub ahead of print.
17. Misbah SA. Rituximab-induced accelerated cryoprecipitation in HCV-associated mixed cryoglobulinaemia has parallels with intravenous immunoglobulin-induced immune complex deposition in mixed cryoglobulinaemia. Arthritis Rheum 2010; 62: 3122.
18. Schiff RI. Transmission of viral infections through intravenous immunoglobulin. N Engl J Med 1994; 15: 1649–1650.
19. Chapel HM, Spickett GP, Ericson D, Engl W, Eibl MM & Bjorkander J. The comparison of the efficacy and safety of intravenous versus subcutaneous immunoglobulin replacement therapy. J Clin Immunol 2000; 20: 94–100.
20. Misbah SA, Baumann A, Fazio R et al. A smooth transition protocol for patients with multifocal motor neuropathy going from intravenous to subcutaneous immunoglobulin therapy: an open label proof-of-concept study. J Peripheral Nerv Syst 2011; 16: 92–97.
Further reading
Castigli E & Geha RS. Molecular basis of common variable immunodeficiency. J Allergy Clin Immunol 2006; 117: 740–746.
Cavazzano-Calvo M & Fischer A. Gene therapy for severe combined immunodeficiency: are we there yet? J Clin Invest 2007; 117: 1456–1465.
Eibel H, Salzer U & Warnatz K. Common variable immunodeficiency at the end of a prospering decade: towards novel gene defects and beyond. Curr Opin Allergy Clin Immunol 2010; 10: 526–533.
Jolles S, Sewell WAC & Misbah SA. Clinical uses of intravenous immunoglobulin. Clin Exp Immunol 2005; 142: 1–11.
Misbah S, Kuijpers T, van der Heijden J et al. Bringing immunoglobulin knowledge up to date: how should we treat today? Clin Exp Immunol 2011; 166: 16–25.
Notarangelo LD, Fischer A, Geha RS et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee – 2009 update. J Allergy Clin Immunol 2009; 124:1161– 1178.