Jason J. Heavner, MD, Jussi J. Saukkonen, MD, and Kathleen M. Akgün, MD
CHAPTER OUTLINE
■ RESPIRATORY FUNCTION
■ COMMON PULMONARY COMPLICATIONS
■ TOBACCO AND NICOTINE
■ MARIJUANA
■ COCAINE
■ AMPHETAMINES AND OTHER STIMULANTS
■ CAFFEINE
■ OPIOIDS
■ ALCOHOL
■ SEDATIVE–HYPNOTICS
■ VOLATILE SUBSTANCES
■ NITROUS OXIDE
■ ANABOLIC STEROIDS
The respiratory tract is an unique interface between the body and the environment; the lungs contain the largest surface area of the body exposed to the external environment (1). The airways and alveoli, which come into close proximity to the vascular bed of the lung, are subject to constant noxious, particulate, and antigenic challenges. The lungs are highly adapted in attenuating such external provocations and in mediating between events at the epithelial and endothelial surfaces. However, a variety of addictive drugs present acute and chronic insults to the respiratory system and can overwhelm the local capacity for recovery (Table 77-1).
TABLE 77-1 DRUGS ASSOCIATED WITH PULMONARY COMPLICATIONS
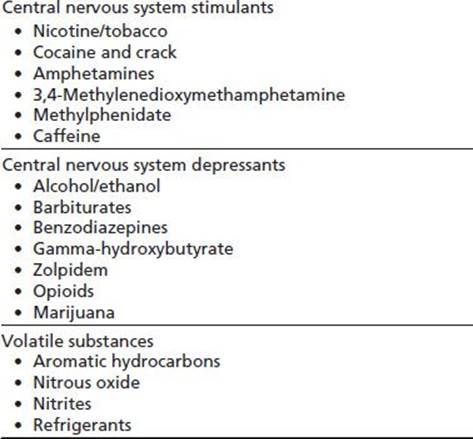
Inhalation, injection, or ingestion of addictive drugs can have adverse effects within the airways, lung parenchyma, and pulmonary vascular bed. Respiratory complications also may arise indirectly from the effects of drugs on the central nervous system (CNS), cardiovascular system, and immune system. This chapter briefly provides an overview of the interactions between the environment and the respiratory tract and then discusses the pulmonary complications associated with several commonly abused drugs.
RESPIRATORY FUNCTION
The principal function of the respiratory tract is gas exchange. Each alveolus is surrounded by an extensive network of thin-walled capillaries; this elaborate network of airways and capillary beds provides a large surface area for gas exchange. The respiratory tract contains and excludes foreign materials and provides an interface for immune sampling of antigens and provides a large surface area for absorption of inhaled substances and drugs. The respiratory tract also detoxifies and metabolizes proteins, drugs, and other potentially injurious substances. Drugs of abuse can derange these critical, interrelated functions of the respiratory tract and lungs.
The cough reflex and the mucociliary escalator provide essential mechanical barriers to foreign matter. At the cellular level, resident alveolar macrophages ingest particles and pathogens and then process, digest, and transport them to lymph nodes for antigen presentation. Large numbers of marginated leukocytes and platelets are stored within the pulmonary vasculature and are mobilized by pulmonary epithelial or endothelial cell injury, causing exudation, inflammation, and compromise of gas exchange (2). Local immune responses recruit other leukocytes, which may further contain or help remove any offending matter through granuloma formation or phagocytosis (2).
Respiration is under extensive neural control and, consequently, is susceptible to the effects of CNS depressants and stimulants. Respiratory automaticity is provided by the medulla oblongata within the brainstem and modulated by the reticular activating system, cerebral cortex, and peripheral sensors (2). Hypoxemic drive comes from the carotid body, which, when stimulated, causes hypotension and bradycardia. Hypoxemic drive is blunted by age, obesity, chronic obstructive pulmonary disease (COPD), and CNS depressants. Pulmonary processes such as interstitial edema, pulmonary emboli, and chemical products of anaphylaxis cause dyspnea and tachypnea through C fiber receptor stimulation (including J receptors in the lung parenchyma). Irritant receptors in the epithelium of the larynx and large airways respond to a variety of chemical irritants and mechanical stimuli, causing cough, broncho-constriction, sneezing, laryngospasm, and mucus secretion. Receptors within the lung and in the brain are responsive to a number of addictive substances (2).
Addictive drugs may affect the lung through direct local inflammation, increased susceptibility to infections, airway reactivity, impairment of pulmonary vascular integrity, acute lung injury, structural injury, and derangements of gas exchange. Polysubstance use is associated with a variety of injuries, making it difficult to ascribe a particular respiratory complication to a single agent. Coexisting pulmonary pathology may worsen the acute and chronic physiologic effects of an addictive drug on the lungs.
COMMON PULMONARY COMPLICATIONS
Several families of drugs cause common respiratory complications, including adverse effects related to respiratory depression, infections, routes of abuse, and contaminants.
Respiratory Depression
With the exception of nicotine, cocaine, and amphetamines/ stimulants, all of the drugs discussed in this chapter inhibit respiration, causing respiratory depression or respiratory failure. Respiratory depression is the inability to maintain normal ventilation. Respiratory failure develops when the lungs cannot maintain normal gas exchange. Loss of protective reflexes, including the gag reflex and the ability to cough, and decreased minute ventilation are commonly seen in respiratory depression associated with drug use. Hypoventilation from respiratory depression contributes to hypoxemia through atelectasis, hypercapnia, and impaired clearance of secretions with potential aspiration events (3). With drug overdose, lethargy may progress rapidly to stupor, severe respiratory depression, coma, and respiratory arrest. With intravenous drug use, death from respiratory failure can occur especially rapidly.
The diagnosis of drug-induced respiratory failure may be obvious from the physical environment, clinical presentation, history and signs of drug abuse, and a positive toxicology screen. A complete chemistry panel, toxicology screen, electrocardiogram, arterial blood gas, and chest radiograph should be performed. Airway protection is paramount, and patients with known or suspected overdose should be admitted to an intensive care unit for monitoring of their respiratory, neurologic, and hemodynamic status (3). Inpatient management of drug overdoses generally consists of administration of intravenous fluids, thiamine, glucose, and naloxone, which may be used both diagnostically and therapeutically. Therapeutic naloxone usually requires continuous infusion due to its short half-life.
Atelectasis
In patients with respiratory depression, shallow respirations result in decreased functional residual capacity, which contributes to airway closure in dependent regions of the lung during expiration (4). Ineffective cough and aspirated oral and gastric secretions, with loss of surfactant, contribute to the development of atelectasis. Unventilated areas distal to closed airways maintain perfusion, resulting in shunting and subsequent hypoxemia. Reversal of hypoventilation with correction of underlying respiratory depression and reexpansion of atelectatic areas of the lung with positive- pressure ventilation, incentive spirometry, chest physiotherapy, respiratory suctioning, and supplemental oxygen generally are indicated. Bronchoscopy may be needed for refractory atelectasis in the face of severe hypoxemia or to rule out an endobronchial lesion or aspirated foreign body (5).
Aspiration Syndromes
The risk of aspirating oropharyngeal or gastric contents into the lower respiratory tract increases with decreasing levels of consciousness (6). The common aspiration syndromes include aspiration pneumonitis, aspiration pneumonia, airway obstruction, and diffuse aspiration bronchiolitis (7). Depending on the type and quantity of material aspirated and the host’s response, the clinical manifestations of aspiration can vary from mild hypoxemia to acute respiratory distress syndrome (ARDS) and death. In patients hospitalized for drug overdose, 10% have aspiration pneumonitis; drug overdose is the most common cause of aspiration pneumonitis (7). Preventive care should be directed toward airway control, maintenance of at least a 30-degree angle, avoidance of oral intake while lethargic, oral hygiene, pulmonary toilet, supplemental oxygen, and antibiotics as indicated. Empiric antibiotic coverage for aspiration in the absence of infiltrates and clinical and laboratory indicators of infection may be counterproductive, merely leading to the selection of nosocomial pathogens (8).
Respiratory Infections
Chronic users of addictive drugs are susceptible to a variety of respiratory infections. Persons who use drugs are at risk of direct infection from a nonsterile injection technique or bacteria-contaminated injected substances. Many drugs have adverse effects on leukocyte function, contribute to malnutrition with resultant immune dysfunction, are injected under unsterile conditions, or may be contaminated with pathogens (9,10). Infectious complications also may be related to coexisting HIV infection, cirrhosis, aspiration, smoking, or inhibition of the mucociliary escalator. Asthma, drug-related bronchospasm, obstructive pulmonary disease, and interstitial lung disease (ILD) may require chronic inhaled or systemic corticosteroid therapy; these immunosuppressant therapies may contribute to infection risk.
The types of respiratory infections in drug users include sinusitis, acute bronchitis, community-acquired and aspiration pneumonias, septic emboli, fungal infections, and mycobacterial infections (7). Patients with pneumonia are generally ill appearing and most often present with acute symptoms of fever, productive cough, dyspnea, and pleuritic chest pain. Polymicrobial and atypical infections make subacute or atypical presentations more common. The chest radiograph may reveal lobar pneumonia, bibasilar or superior segment lower-lobe infiltrates suggestive of aspiration, or nodular infiltrates in the lower lung fields consistent with aspiration (11). Sputum Gram stain can be helpful to confirm lower respiratory infection and indicate whether the specimen is of sufficient quality to make the accompanying sputum culture reliable. Sputum acid-fast staining and culture should also be performed if there is suspicion for mycobacterial infections. Blood cultures are warranted in many patients, especially those who are toxic, ill appearing, immunocompromised, or otherwise high risk (e.g., alcohol abuse, intravenous drug abuse). Early and appropriate administration of antibiotics tailored to the suspected pathogen is essential (11).
Respiratory Complications of Contaminants
Illicit drugs vary greatly in purity. Adulterants, pharmaco-logically active substances used to increase quantities of the drug of interest or to enhance drug delivery, are frequently identified in illicit drugs. Commonly used adulterants include mannitol and other sugars, cellulose, and talc, as well as other drugs such as phenobarbital, fentanyl, methaqualone, caffeine, procaine, noscapine, and levamisole (12,13). Additionally, drugs can be contaminated with manufacturing by-products such as lead, aluminum, and glass. The lungs act as a filter, trapping inhaled or injected foreign substances that may incite local inflammatory or fibrotic responses. Contaminating microorganisms such as Aspergillus in marijuana may lead to hypersensitivity responses or to pulmonary infection, particularly in immu-nocompromised hosts (14–16). A 2011 review identified Bacillus and Clostridium species to be the most common microbiologic contaminants (12).
The role of herbicides such as paraquat in contaminating inhaled drugs and causing pulmonary fibrosis is controversial. While epidemiologic evidence of paraquat-induced lung injury is lacking, paraquat is a well-known cause of occupational lung disease in workers with exposure (17). Paraquat has also been associated with fatal acute pneumonitis when used to alter the taste of alcohol in illicit alcohol users (18).
Occupational Lung Disease
Agricultural workers may develop pulmonary complications related to cultivation of certain plants for the drug industry (19). Drug-related lung disease among agricultural workers in the drug industry may be underappreciated because of the illicit nature of an industry that is not subject to occupational safety regulations or to exposure mitigation regimens. Pneumoconiosis, herbicide-related interstitial fibrosis, antigen-induced hypersensitivity pneumonitis, and nicotine poisoning from green tobacco sickness are among the occupational pulmonary hazards for agricultural workers (19). Furthermore, organophosphate poisoning can also affect agricultural workers, resulting in bronchospasm and respiratory depression (20).
Respiratory Complications of Injected Drugs
Opioids, stimulants, and combinations thereof are commonly injected into the veins. The resulting pulmonary complications may be acute or chronic. Acute problems are likely to be severe, including respiratory failure and acute pulmonary edema (3). Chronic pulmonary problems include the development of interstitial and bullous lung disease, endovascular and respiratory infections, pulmonary hypertension, tuberculosis, and cancer (3,21,22). Potential pulmonary complications associated with the drug, its contaminants, or the route of administration include the following conditions.
Talc Granulomatosis
Talc (magnesium silicate) is widely used as a filler in medications such as buprenorphine, oxycodone, and methylphenidate. These drugs may be crushed and injected or may be used to adulterate other inhaled and injected drugs (23). A syndrome similar to sarcoidosis may result, with insidious onset of granulomatous interstitial fibrosis (24). Dyspnea, particularly with exertion, and cough are the most common symptoms. The retina should be examined in all patients in whom the diagnosis is being considered because talc retinopathy occurs in more than half of patients with pulmonary manifestations (23).
The chest radiograph is normal in up to half of patients with talc-related fibrosis (24). A diffuse micronodular interstitial infiltrate may be evident, particularly in the mid-lung zones. Nodules can coalesce to opacify entire lobes as the disease progresses. High-resolution computed tomography may reveal diffuse ground glass opacity and confluent perihilar masses, the latter containing areas of high attenuation consistent with talc (24). Pulmonary function tests (PFTs) typically reveal a low diffusion capacity before any other abnormality (25,26). Bronchoalveolar lavage may demonstrate local lymphocytosis and birefringent intracellular or free talc (23). Lung biopsy may be required to establish the diagnosis, based on histologic changes of granulomas, mononuclear inflammatory cells, lymphocytes, and fibrosis (23). In advanced stages or if there is associated granuloma-tous pulmonary arterial occlusion, pulmonary hypertension and right ventricular failure can occur (27). Patients with progressive symptoms and worsening chest radiograph or PFTs should be given a trial of systemic steroids, although results are variable and unpredictable.
Pulmonary Hypertension
Intravenous drug users may develop chronic pulmonary hypertension from multiple mechanisms, including chronic hypoxemia related to ILD and vasoconstriction, pulmonary embolization of particulate matter from crushed tablets used for injection, pulmonary arterial thrombosis at sites of foreign body granulomatosis, thromboembolic disease, and pulmonary arterial hypertension from the drug itself (e.g., cocaine) or from an adulterant (e.g., levamisole) (13,28). Injection drug use can lead to HIV infection, which is also associated with pulmonary arterial hypertension (29). Acute, reversible pulmonary hypertension has been associated with injection of sympathomimetics, although this may be due to transient decreased left ventricular function and not pulmonary arterial hypertension itself (30).
The most common presentation of pulmonary hypertension is dyspnea on exertion (31). Physical examination and electrocardiogram may be normal or may be consistent with right ventricular enlargement and failure. Treatment options include supplemental oxygen for patients who are hypoxemic, anticoagulation for patients at increased risk for venous thromboembolism, and diuretics for patients with peripheral edema. Patients should be referred to a pulmonary hypertension center for hemodynamic evaluation and consideration of advanced therapy (31). Harm reduction interventions such as syringe filters may reduce pulmonary embolization of particulate matter and subsequent pulmonary hypertension in injection drug users (32–34).
Septic Thromboemboli
Septic pulmonary embolism is a common pulmonary complication among intravenous drug users and may result from tricuspid endocarditis or from infected injection-site thrombophlebitis (35). The most common reason for hospitalization in intravenous drug users is cutaneous injection-related infections; 9% of these admissions are complicated by tricuspid valve endocarditis (35,36). The organism most frequently isolated from sputum or blood cultures is Staphylococcus aureus (35,37).
Patients typically present in an acute toxic state with fever, dyspnea, chest pain, and leukocytosis. Radiographic examination of the chest may reveal bilateral necrotizing infiltrates or single or multiple pulmonary nodules, which frequently cavitate (38). The lesions may coalesce to form large cavities that communicate with a bronchus (38). Infections may also be complicated by bronchopleural fistulas, empyema, or pneumothorax (37).
Therapy with prolonged courses of antibiotic therapy and, in some cases, invasive drainage or excision can result in complete resolution (38,39). However, the mortality associated with septic thromboemboli is between 10% to 20%, and residual pleural scarring and fibrosis are common among survivors (35,37,39).
Drug and Needle Embolization
Occasionally, needles may break off inadvertently during injection, or entire needles may embolize if left in place after injection (40). This situation is more likely to occur when less accessible injection sites are used. Chest imaging may demonstrate needle fragments within chest soft tissue or lodged within the pulmonary vasculature. No long-term complications have been reported after conservative management; therefore, no specific therapy is necessary for needle emboli to the lung (40). Embolization of particulate matter after injection of crushed drugs, such as Percocet, can result in cardiopulmonary arrest (41).
Pneumothorax
Pneumothorax, unilateral or bilateral, may develop from inadvertent puncture of the lung during attempted needle injection into a jugular or subclavian vessel (42). This condition also may result from cavitating septic thromboemboli (37). Large pneumothoraces, those associated with hypoxemia or respiratory failure, or those with concomitant empyema, require tube thoracostomy (43).
Empyema
Cavitating infections from septic emboli, pneumonia, or unclean needles may contaminate the pleural space and lead to empyema (35,38). Empyema requires pleural drainage with tube thoracostomy or surgical debridement.
Mycotic Aneurysms
Septic emboli may lead to the development of mycotic aneurysms of the pulmonary vasculature (44). Patients may present with evidence of endovascular infection, with or without hemoptysis. Contrast-enhanced computed tomography demonstrates nodular lesions associated with vasculature. If there is massive hemoptysis, this condition may be fatal despite endovascular or surgical intervention. Patients without hemoptysis may be successfully treated with antibiotic therapy (44,45).
Hemothorax
Rupture of a subclavian aneurysm created by multiple injections has been reported to cause a massive hemothorax (46).
Pulmonary Emphysema
Bullous emphysema may develop in intravenous drug users, in association with talc granulomatosis or idiosyncratically (24,47). The mechanism is unknown.
Respiratory Complications of Inhaled Drugs
Inhalation has become a preferred route for use of many addictive drugs. Inhalation of a drug requires minimal supplies and provides a rapid onset of action, dose minimization, avoidance of intravenous injection, and avoidance of hepatic first-pass metabolism. Drugs may be inhaled nasally (“snorting”), rolled as cigarettes, sprinkled into smoked tobacco, smoked through pipes or vaporizers, or inhaled orally. The route of inhalation may affect the dose delivered and the onset of action. Deep inhalation or smoking may result in an onset of action within seconds, whereas nasal inhalation has a slightly delayed onset, measured in seconds to minutes (48). Various delivery devices and techniques have been developed to control dose and effects, to provide convenience, and to incorporate cultural, behavioral, and aesthetic elements. For example, water pipes, used for smoking a variety of drugs throughout the world, are associated with a café culture and have an aesthetic design element. More recently, detrimental effects of water pipe smoking comparable to those of cigarettes have been identified, including acute and chronic expiratory airflow obstruction and marked increases in carboxyhemoglobin (49–51).
Inhaled drugs include fine powders, smoked material, gases or other volatiles, and combinations of these drugs. Inhaled powders are heterodisperse, varying considerably in size, with a geometric standard deviation greater than 2 (2). The momentum of inhaled particles greater than 6 μm causes them to impact proximally against convoluted upper airway walls and bifurcating large airways of the lung where they may be absorbed. Smaller particles (1 to 5 μm) are carried by the airstream to the distal airways, where they are deposited chiefly by sedimentation, settling as a result of gravitational forces (2).
Inhalation of talc or other fibrogenic substances may lead to the development of granulomatous inflammation or fibrosis, as discussed previously in this chapter. Smoke consists of gas and particulate phases, including carbon monoxide, oxidants, aldehydes, alcohols, nitrosamines, benzene derivatives, and other inorganic and organic substances, many of which cause mucosal injury and inflammation (52).
Chronic Bronchitis and Emphysema
These conditions are commonly associated with smoking tobacco. Chronic bronchitis and emphysema may also be associated with chronic use of marijuana, cocaine, and other drugs (53). Cough, dyspnea, and mucus hypersecretion are found in individuals with chronic bronchitis. Destruction of lung parenchyma, with resultant pulmonary emphysema, may occur with inhaled tobacco, marijuana, and chronic opium use (49,54,55). Increased susceptibility to airway reactivity and acute bacterial bronchitis may also be seen (53).
Bronchospasm
Airway reactivity often is seen with inhaled heroin, cocaine, tobacco, and marijuana (56,57). Patients typically present with dyspnea, tachypnea, tachycardia, and wheezing, beginning within minutes to hours after inhalation. Early arterial blood gases, if obtained, may demonstrate respiratory alkalosis, which may progress to hypercapnia and respiratory acidosis. The chest radiograph typically is clear although hyperinflation may be present. Patients are treated with supplemental oxygen, bronchodilators, steroids, and, if severe, mechanical ventilation. Patients with new-onset asthma should be assessed for use of inhaled drugs (58).
Barotrauma
Inhalation of cocaine, heroin, 3,4-methylenedioxymetham-phetamine, marijuana, tobacco, and volatile substances is associated with barotrauma, including pneumothorax and pneumomediastinum (59). Extreme breath holding against a closed glottis (a prolonged Valsalva maneuver) is sometimes used in an attempt to increase drug effect. This maneuver results in high negative intrathoracic pressure and hyperinflation that may result in alveolar bleb rupture with dissection of air along peribronchial paths into the mediastinum, pleural cavities, skin, and retropharyngeal space (60). Some users exhale smoke forcefully into another user’s mouth, causing markedly elevated positive airway pressure and potentially barotrauma as well as transmission of respiratory pathogens (61). Patients typically present with acute chest or back pain and dyspnea, with or without hypoxemia (55,62,63).
Hemoptysis
Hemoptysis results from mucosal irritation or ulceration anywhere within the respiratory tract (i.e., epistaxis, sinusitis, or bronchitis), from pulmonary infarction, or from diffuse alveolar hemorrhage (64,65).
Asphyxiation
Intentional inhalation of halogenated hydrocarbons, such as 1,1-difluoroethane (electronics cleaning gas), is associated with sudden death from asphyxiation (66). The mechanism of hypoxemia is thought to be from the loss of partial pressure of oxygen in the inspired air as well as from destruction of the alveolar basal membrane resulting in acute noncardiogenic pulmonary edema (67).
TOBACCO AND NICOTINE
Within the lung, smoking has profound effects, altering the immunologic and structural milieu. Cigarette smoke contains more than 4,500 components that are associated with a high rate of lung cancer, COPD, bronchitis, and airway reactivity (52). Several factors determine the effects of tar and other pyrolysis products on the lung. Contributing factors include individual susceptibility to the various adverse effects of smoking, heterozygosity for the gene that causes alpha-1-antitrypsin deficiency, the quantity of cigarettes smoked, years spent smoking, and the manner in which they were smoked (e.g., the depth of inhalation) (49,68).
Tobacco smoke stimulates chemotactic cytokines leading to neutrophil and monocyte migration and cell mediators that cause epithelial injury and permeability changes (69). Pulmonary leukocytes are activated by the local, intrapul-monary cytokines (including interleukin-8, tumor necrosis factor alpha, and interleukin-1) and by epithelial cells, fibroblasts, and leukocytes. Neutrophils and macrophages release serine proteases, reactive oxygen species, matrix metalloproteinases, and other potentially injurious cell products. Imbalance between proteases and antiproteases is thought to play a role in the development of local injury (69,70). Elastin and collagen degradation develops, resulting in loss of pulmonary architecture and progressive loss of functional gas-exchanging units. T cells and eosinophils also play a pathogenic role after long-standing injury (69).
Smoking is associated with acute and chronic lower respiratory tract infections (bronchitis and pneumonia) and obstructive and restrictive lung diseases. Smokers have a more rapid decline in forced expiratory volume in one second (FEV1) than do nonsmokers, which may lead to symptoms such as dyspnea on exertion and fatigue (71). Chronic airflow obstruction is common and, when associated with persistent hypoxemia, may lead to pulmonary hypertension, cor pulmonale, and right heart failure (72). Cigarette smoking is an important risk factor for lung cancer. Cigarette smoking is also a risk factor for ILDs, identified by restrictive physiology on PFTs (72,73). Smoking cessation may slow the rate of decline of lung function, attenuate symptoms, lower the risk of developing lung cancer, and decrease the incidence of lower respiratory infections (71).
Environmental tobacco smoke has become a major public health issue (74). In children, environmental tobacco smoke contributes to lower respiratory illnesses, chronic respiratory symptoms, middle ear disease, reduced lung function, childhood asthma, and wheezing in early childhood (75). In adults, environmental tobacco smoke appears to increase the risk of lung cancer, cardiovascular diseases, and acute respiratory symptoms and illnesses (74,76,77).
Smoking cessation may slow the rate of decline of lung function, attenuate symptoms, lower the risk of developing lung cancer, and decrease the incidence of lower respiratory infections (78). Nicotine-based and non–nicotine-based medications can improve successful smoking cessation (79,80). Development of novel drug delivery routes such as inhaled medications is likely to improve successful quit attempts (80,81). Electronic cigarettes may have a role in successful smoking cessation though the long-term safety has not been proven (82,83).
The following are the clinical syndromes associated with the use of tobacco.
Chronic Obstructive Pulmonary Disease: Chronic Bronchitis and Pulmonary Emphysema
COPD is characterized by a persistent airflow limitation that is usually progressive (70). COPD is associated with a chronic inflammatory response in the airways and the lung. In smokers above the age of 45, 40% to 50% meet criteria for the diagnosis of COPD (84,85). COPD is diagnosed using spirometry according to the 2013 GOLD guidelines. Fixed airway obstruction is defined as FEV1/forced vital capacity (FVC) less than 70% after bronchodilator administration (70). Use of a fixed ratio to diagnose COPD does not account for normal changes in airflow limitations with age. Therefore, the American Thoracic Society and the European Respiratory Society recommend COPD diagnosis as FEV1/FVC below 5% of the lower limit of normal for healthy controls (86). COPD was the third leading cause of death in the United States from 2007 to 2012 (84,85). The pathogenesis of COPD is associated with oxidative stress, protease–antiprotease imbalance, and cytotoxic lymphocytic inflammation (70).
Clinically, patients typically present with dyspnea, wheezing, chronic cough or chronic, nearly daily sputum production. Chronic bronchitis is sputum production for at least 3 months in 2 successive years in the absence of other causes of chronic cough. Airway changes in chronic bronchitis are nonspecific, but consist of mucus gland hypertrophy in intermediate-sized airways. Overproduction of mucus may overwhelm the mucociliary escalator, which is compromised by tobacco smoke (87). Concurrent airway reactivity is often described.
Pulmonary emphysema results from irreversible enlargement of the airspaces distal to the terminal bronchiole with destruction of the alveolar wall. Interstitial fibrosis may occur but is not a major pathologic finding. Centrilobular emphysema in the upper portions of the lungs, as well as peripheral paraseptal emphysema, is commonly found. The latter is associated with the development of large bullae. Hyperinflation and flattening of the diaphragm lead to a mechanical disadvantage for the contractility of the major respiratory muscle (88). Gas exchange abnormalities are seen because of hyperinflation and associated mechanical disadvantage, loss of surface area, and suboptimal ventilation–perfusion matching, which contributes to impaired oxygenation and carbon dioxide retention. There may be associated chronic bronchitis or airway reactivity, or both. Pneumothorax may develop from rupture of a subpleural emphysematous bleb.
In addition to the clinical diagnostic findings of dyspnea, cough, or sputum with evidence of obstruction on spirometry, PFTs may also reveal hyperinflation, air trapping, and decreased diffusion capacity. Radiographic imaging may show emphysematous destruction of the lung parenchyma. Over time, pulmonary hypertension and right heart failure can develop. Mainstays of treatment include smoking cessation, bronchodilator therapy, inhaled or systemic steroids, antibiotics for exacerbations supplemental oxygen therapy, and pulmonary rehabilitation (70).
Airway Reactivity
Smoking tobacco promotes bronchial hyperreactivity, as measured by methacholine bronchoprovocation studies, even in patients without obstructive lung disease (89). Animal studies suggest that early smoke-induced bron-choconstriction is caused by nicotine, and it is mediated by cholinergic pathways and by tachykinin release from bronchopulmonary C fibers (90). A delayed and more sustained bronchoconstrictive response is induced by nonnicotinic components of tobacco smoke that elicit the release of the eicosanoids, thromboxane A2, prostaglandin D2, and prostaglandin F2 alpha, acting on airway smooth muscles (91,92). Smokers also have increased numbers of activated leukocytes, which may contribute to asthmatic symptoms (93,94). In some individuals with severe airflow obstruction, there are correlations between total serum immunoglobulin E (IgE) levels and an impaired response to corticosteroids, which may contribute to reactive airway symptoms in non-asthmatics or exacerbate symptoms in patients with preexisting asthma (69).
Patients typically complain of cough, dyspnea especially on exertion, and chest tightness. PFTs may show an obstructive pattern with or without a bronchodilator response. Bronchodilator therapy and inhaled corticosteroids are the mainstays of therapy (95).
Interstitial Lung Disease
In addition to obstructive lung disease, smoking is associated with an increased risk of ILDs (96). Cigarette use is the primary risk factor for respiratory bronchiolitis–associated ILD (RB-ILD), desquamative interstitial pneumonia (DIP), and pulmonary Langerhans cell histiocytosis (PLCH) (96). Cigarette smoking is also associated with idiopathic pulmonary fibrosis, rheumatoid arthritis–associated ILD, acute eosinophilic pneumonia (AEP), and Goodpasture syndrome (96). In patients with anti–glomerular basement membrane antibodies with glomerulonephritis, 100% of smokers developed diffuse alveolar hemorrhage while only 20% of nonsmokers had pulmonary hemorrhage (97). Underlying pathogenesis includes bronchiolar inflammation, increased interstitial macrophages, and increased transforming growth factor–beta production, which modulates the pulmonary fibrotic response (96). Patients present with an insidious progression of dyspnea, dry cough, diffuse infiltrates, restrictive physiology on PFTs, and impaired diffusion capacity (98). Chronic obstructive lung diseases are often concurrent with ILDs in smokers (96).
Surgical lung biopsy is needed to reliably diagnose ILDs (98). However, based on the results of high-resolution computed tomography and clinical features, a clinicora-diographic diagnosis can be made, particularly at medical centers specializing in ILD with expert chest radiologists and ILD pulmonologists (96). Bronchoscopic biopsies and bronchoalveolar lavage are useful in the diagnosis of PLCH and AEP. In RB-ILD, DIP, and PLCH, smoking cessation is a critical component of management (96). Smoking cessation alone may lead to stabilization of symptoms and improvement in radiographic and functional studies. Pharmacologic management involves a trial of systemic corticosteroids and other immunomodulatory agents (98).
Pulmonary Hypertension and Cor Pulmonale
Chronic hypoxic vasoconstriction of the pulmonary vasculature leads to pulmonary arterial hypertension and right heart failure (31). Pulmonary hypertension complicates at least 20% of patients with COPD (99). Symptoms include dyspnea and fatigue. As the disease progresses, patients also experience palpitations and syncope (31). On physical examination, clinicians may find tachycardia, prominent neck veins, tricuspid insufficiency murmur, prominent P2 heart sound, hepatojugular reflux, ascites, and peripheral edema. Screening for pulmonary hypertension is performed by transthoracic echocardiography. Right heart catheterization is necessary to confirm the diagnosis (31).
The primary therapy for hypoxemia-related pulmonary hypertension consists of optimizing treatment of the cause of the hypoxemia (31). Supplemental oxygen for at least 18 hours/day prolongs survival and improves symptoms. Additional options for primary therapy include anticoagulation for patients at increased risk for venous thromboembolism and diuretics for patients with edema. At the present time, there are no recommendations for the use of pulmonary arterial hypertension therapies, such as phosphodiesterase inhibitors, intravenous prostanoids, and endothelin receptor antagonists in patients with cor pulmonale. Patients should be referred to a pulmonary hypertension center for hemodynamic evaluation, consideration of advanced therapy, and clinical trial enrollment (31).
Pneumothorax
Primary spontaneous pneumothorax typically occurs in thin, tall, young men, with a 22-fold increased relative risk in smokers (100). Secondary spontaneous pneumothorax may occur after rupture of a subpleural bleb in patients with bullous emphysema. Occasionally, cavitating infections or bronchogenic carcinomas also may cause pneumothorax (101).
Hemoptysis
Most commonly, hemoptysis results from acute bronchitis. However, in smokers, the presence of an endobronchial tumor must be considered. Additionally, 100% of smokers with glomerulonephritis from anti–glomerular basement membrane antibodies developed diffuse alveolar hemorrhage, while this is only seen in 20% of nonsmokers (97).
Lung Cancer
Lung cancer is the leading cause of cancer death in the United States. Worldwide, cigarette smoking is responsible for more than 80% of lung cancers (102). It is estimated that the risk of lung cancer is increased 21% in adult nonsmokers exposed to second-hand tobacco smoke compared to those with no second-hand smoke exposure (74). Approximately 70% of lung cancers present in advanced stages, leaving most patients to have unresectable disease with a 5-year survival rate of less than 20% (102).
Patients may present with asymptomatic, incidentally noted pulmonary nodules or with weight loss, cough, chest or bone pain, fatigue, hoarseness, superior vena cava syndrome, or hemoptysis. Diagnosis may be made by needle or bronchoscopic cytologic specimens, endobronchial biopsy, transbronchial biopsies, endobronchial ultrasound-guided mediastinal needle aspiration, or mediastinoscopy, or with surgical resection. Treatment may be definitive, in the case of a completely resected, margin-free solitary pulmonary nodule, or it may include radiation or chemotherapy, depending on the cell type and stage.
Early detection is paramount for increased survival. Recently, the National Lung Screening Trial showed decreased mortality in high-risk smokers (smoked within the past 15 years and at least a 30-pack-year smoking history) with the use of low-dose spiral computed tomography lung cancer screening (103).
Compared to nonsmokers, smoking cessation decreases the relative risk for lung cancer mortality from 23.4 to 5.3 in men and from 21.1 to 2.4 in women after 15 years of abstinence (104). For patients who are unable to quit, smoking reduction also appears to decrease the likelihood of lung cancer, albeit to a lesser extent (105).
Hypersensitivity Pneumonitis
A variety of molds may be present on tobacco plants that then are inhaled by workers harvesting the crop or, theoretically, by smokers (106,107). In some individuals, these molds, including Aspergillus and thermophilic actinomycetes, may induce hypersensitivity reactions on exposure. Workers typically experience improvement of symptoms when away from work. Clinical features include cough, fever, and obstructive and restrictive findings on PFTs (106,107). With prolonged exposure over years, pulmonary fibrosis may develop (106). Although smoking can show serologic evidence of exposure, it is unclear whether hypersensitivity pneumonitis develops in smokers (108).
Lipoid Pneumonia
The rare complication of lipoid pneumonia has been reported among chewers and smokers of blackfat tobacco in Guyana, where petroleum jelly is applied to tobacco leaves to moisturize them and to enhance their flavor (109). Patients may present with cough and a localized infiltrate, which may suggest chronic pneumonia or bronchogenic carcinoma (110). The density of the lesion on computed tomography scan usually suggests the diagnosis (110).
Water Pipes
Although considered by the public to be less harmful than cigarette smoking, water pipe use is likely to be a cause of COPD (51). One 30-minute water pipe tobacco smoking session is associated with increased carboxyhemoglobin levels, increased systolic and diastolic blood pressures, increased heart and respiratory rates, and decreases in small airway forced expiratory flow and peak expiratory flow rates (50). Over time, continued water pipe smoking is associated with a decreased FEV1/FVC and FEV1; these declines are statistically similar to those in cigarette smokers (51).
Electronic Cigarettes
The electronic cigarette (e-cigarette) is becoming a common inhaled nicotine delivery device of nicotine vapor. By 2011, about 20% of adult cigarette smokers had used electronic cigarettes, and approximately 6% of all adults (smokers and nonsmokers) have tried e-cigarettes (111). Analyses from the U.S. FDA have found numerous carcinogens in e- cigarettes, including nitrosamines and diethylene glycol (112). While long-term health studies are lacking, short-term use has been shown to increase both bronchial inflammation and lung flow resistance (113).
MARIJUANA
Marijuana may be ingested or smoked as cigarettes or through water pipes of various designs. The smoke is qualitatively similar to that of tobacco, but it lacks nicotine and contains tetrahydrocannabinol and other cannabinoids (114,115). Smoking marijuana results in inhaling three times more tar than cigarettes and causing a fivefold higher carboxyhemoglobin level in the blood than cigarettes (115). Alveolar macrophages from marijuana smokers do not phagocytose properly, have decreased ability to kill bacteria and tumor cells, and produce decreased amounts of nitric oxide and cytokines, including tumor necrosis factor alpha, granulocyte–macrophage colony-stimulating factor, and interleukin-6 (116). Marijuana smoke increases oxidative stress within the lung, causing glutathione stores to be depleted within alveolar macrophages (117). While earlier studies have conflicting results, more recent data demonstrate that long-term marijuana use is associated with an increased risk of lung cancer (21,118). Heavy marijuana use is also associated with laryngeal cancer (119).
Chronic Obstructive Pulmonary Disease
Marijuana smokers may suffer some of the same complications as those who smoke tobacco, including COPD and bullous emphysema (21,54,55). Marijuana smokers inhale more tar because the marijuana cigarettes are not filtered, inhalation tends to be significantly deeper, and use is associated with prolonged breath holding (115). However, COPD does not develop consistently. Low cumulative use of marijuana is associated with an increase in FEV1 when spirometry is compared over a 20-year follow-up period (120). Likewise, short-term use is associated with bronchodilation (121). Heavy or long-term use is associated with reversal of improved FEV1 and an increase in COPD symptoms (55,120,121).
Pathogen-Associated Complications
Contaminating microorganisms such as Aspergillus in marijuana may lead to pulmonary infection. This is particularly concerning in immunocompromised hosts (15,16). This association may become more important as increasing numbers of immunocompromised patients turn to medicinal uses of marijuana. Contaminated marijuana has also been associated with fungal hypersensitivity reactions (14).
Lipoid Pneumonia
Respiratory failure because of lipoid pneumonia with pulmonary alveolar proteinosis in a patient with a cadaveric renal transplant has been linked to smoking weed oil prepared from marijuana (122).
Hemp Worker’s Lung
Nonsmoking workers in the hemp industry display evidence of chronic cough and byssinosis, an occupational asthma-like syndrome, and an accelerated decline in FEV1 (123). Much of the cultivation of hemp is an industry separate from that of marijuana production, with differing processing techniques. It is not known whether workers cultivating marijuana for consumption develop a byssinosis-like syndrome.
COCAINE
Cocaine blocks norepinephrine and serotonin reuptake and causes release of norepinephrine, serotonin, and dopamine (124). Cocaine also has local anesthetic effects, blocking sodium and potassium channel flux, thereby reducing action potentials and inhibiting conduction of nerve impulses. Cocaine crosses the blood–brain barrier and stimulates the CNS where, in addition to the well-known effects on the limbic system, it can increase respiratory rate (124).
Cocaine is inhaled nasally, smoked, injected, and ingested. Approximately 20% to 30% of the inhaled dose actually reaches the lung (125). Cocaine often is combined with nicotine, marijuana, and heroin (126). Freebasing is the practice of using volatile solvents to convert cocaine from a salt to a base and to remove adulterants. This potentially incendiary chemical process can lead to extensive cutaneous and inhalational burns. The final freebase product is highly potent and has a rapid onset of action, and, therefore, it is likely to induce pulmonary, cardiac, neurologic, and other complications (127).
Cocaine may induce injury through vasoconstriction, by impairing the integrity of the pulmonary capillary bed, and by cardiovascular effects on the pulmonary vasculature (101). Cocaine is a highly potent bronchoconstrictor when inhaled (128). Additionally, it has prominent effects on vasculature, causing vasoconstriction and permeability changes (101,128). Reduction in diffusion capacity and increased clearance of inhaled technetium-99 compared with nonsmokers are seen in crack cocaine smokers, reflecting damage to the alveolar–capillary membrane, similar to the abnormality associated with smoking tobacco (129). Cocaine also causes alveolar hemorrhage and noncardiogenic pulmonary edema (101).
Cocaine has effects on the immune system, which may contribute to infections or to local inflammatory reactions. It impairs the function of natural killer cells as well as B and T lymphocytes (130,131). Alveolar macrophages from cocaine users have marked defects in their ability to kill bacteria and tumor cells because of a defect in nitric oxide production (116). After in vivo inhalation or injection of cocaine, neutrophils are activated and have enhanced production of interleukin-8, which has been implicated in a number of inflammatory lung disorders, including ARDS (132).
Cocaine may cause a wide range of acute and chronic pulmonary complications through direct mucosal injury and systemic effects. However, the true incidence of cocaine-associated lung diseases is not known. Nonpulmonary complications of cocaine use include primary neurologic events such as seizure or stroke, which may be complicated by pulmonary complications of atelectasis, aspiration, or neurogenic pulmonary edema (133). Pulmonary symptoms commonly associated with cocaine use, particularly use of crack cocaine, include cough productive of carbonaceous sputum, pleuritic chest pain, wheezing, dyspnea, and hemoptysis (133).
Barotrauma
Barotrauma is common with crack cocaine inhalation and is associated with prolonged and forceful deep inhalation, Valsalva maneuver, or “shotgunning” (forceful exhalation of crack smoke into another individual’s respiratory tract) (134). Cocaine-induced pneumomediastinum is considered to be a benign condition in the absence of esophageal or tracheal perforation (135).
Upper Airway Complications and Hemoptysis
A variety of upper airway complications are associated with inhaled cocaine, primarily burns and mucosal irritation or inflammation. The latter may result from the vasoconstrictive properties of cocaine and can cause nasal septal perforation, sinusitis, epiglottitis, and upper airway obstruction (134,136,137). Aspiration of the nasal septum has been reported (138). A vasculitis resembling granulomatosis with polyangiitis in the nasal cavity has been reported in association with nasal inhalation of cocaine (139).
Hemoptysis may result from mucosal irritation or ulceration anywhere within the respiratory tract (i.e., rhinitis, sinusitis, and bronchitis), from pulmonary infarct, or from diffuse alveolar hemorrhage (134). A vasculitis resembling Wegener granulomatosis in the upper airway has been reported in association with nasal inhalation of cocaine (139).
Bronchitis
Both acute and chronic bronchitis may develop from mucosal irritation, as described earlier (134). Often, there is concomitant tobacco use that contributes, in large measure, to COPD.
Airway Burns
Thermal injury is most often secondary to inhalation of smoke or from intratracheal combustion of solvents related to freebasing (140). The anesthetic properties of cocaine make individuals more susceptible to airway burns. Upper airway burns are more common. Lower airway burns may lead to tracheal or bronchial stenosis if the damage is extensive or circumferential (140).
Bronchospasm
Cocaine inhalation, but not injection, causes measurable and clinically significant bronchospasm in asthmatic as well in nonasthmatic, nonatopic individuals (140). Wheezing is reported in 30% to 35% of crack cocaine users during the period of use (140). Cocaine-induced bronchoconstriction is thought to be mediated by airway irritant receptors (57). Cocaine inhalation may precipitate life-threatening exacerbations of asthma requiring intubation and mechanical ventilation (134,141). Bronchospasm tends to be more severe among cocaine users, and cocaine use is associated with inhaled corticosteroid noncompliance and recrudescence of symptoms (56,58,142).
Diffuse Alveolar Hemorrhage
In an autopsy study of cocaine-related deaths, 85% of cases demonstrated alveolar hemorrhage (140). The mechanism of injury is likely to be from damage to the alveolar– capillary membrane. Occult hemorrhage is the most common manifestation (140). Cocaine inhalation may incite alveolar hemorrhage in patients with Goodpasture syndrome (143). Additionally, 70% of cocaine in the United States is contaminated with levamisole—an antihelmintic agent (144). Levamisole-contaminated cocaine is associated with an antineutrophil cytoplasmic antibody (ANCA)-mediated vasculitis. This ANCA vasculitis can result in a pulmonary capillaritis with resulting diffuse alveolar hemorrhage (144).
Pulmonary Edema
Cocaine may induce both noncardiogenic and cardiogenic pulmonary edema. There are several etiologies responsible for noncardiogenic pulmonary edema. Cocaine directly causes increased endothelial permeability of the pulmonary capillaries. Negative pressure pulmonary edema results from high negative intrathoracic pressures during drug inhalation. Neurogenic pulmonary edema can develop from sympathetic-mediated pulmonary venous constriction (140,145).
Cardiogenic pulmonary edema occurs through coronary vasospasm-induced myocardial ischemia or infarction, through arrhythmia, or from acute heart failure related to abruptly increased afterload (134,146). Intravenous cocaine has a chronotropic effect on the heart and may have negative inotropic effects in laboratory administration of cocaine in canines but does not cause an increase in pulmonary capillary wedge pressure (147). These effects may result in cardiogenic pulmonary edema.
Pulmonary Vascular Disease and Infarction
Cocaine use is associated with massive drug-induced pulmonary artery vasoconstriction and may even lead to pulmonary infarction in case reports (148,149). Vasoconstriction, platelet aggregation, vascular damage, and induction of endothelin 1 release may contribute (28). Acute, reversible pulmonary hypertension has been associated with injection of sympathomimetics, although this may be due to decreased left ventricular function and not pulmonary arterial hypertension itself (30). Nuclear ventilation–perfusion scans show areas of mismatch defects within the lung.
Levamisole is commonly used as an adulterant to cocaine and is metabolized to aminorex (13,150). Aminorex, which has been used previously as an anorexic, can cause fatal pulmonary arterial hypertension when used with cocaine (150). Intravenous cocaine use is associated with talc granulomatosis, and when associated with granulomatous pulmonary arterial occlusion, pulmonary hypertension and right ventricular failure can occur (27). One autopsy series of cocaine-related deaths reported pulmonary artery medial hypertrophy in the absence of talc or other debris in 20% of cases (151).
Patients may present with symptoms suggestive of pulmonary embolism, pleuritic chest pain, dyspnea, and hypoxemia and uniformly present with exertional dyspnea (29). Nuclear ventilation–perfusion scans show areas of mismatch defects within the lung.
Eosinophilic Hypersensitivity Pneumonitis (“Crack Lung”)
Crack cocaine use has been associated with a mild Loeffler syndrome, with transient migratory pulmonary infiltrates and eosinophilia (152). The condition is usually self-limited and may not require treatment (152). A more severe reaction may occur 1 to 48 hours after heavy cocaine smoking, which consists of chest pain, cough with hemoptysis, dyspnea, bronchospasm, pruritus, fever, diffuse alveolar infiltrates, and pulmonary and systemic eosinophilia (153,154). Elevated circulating IgE may be found. Histopathologically, the syndrome is most consistent with AEP with extensive IgE deposition (154). This syndrome may represent an IgE-dependent hypersensitivity response with mast cell degranulation, eosinophil recruitment, and tissue damage (154). Recurrent episodes may occur with continued cocaine inhalation. It is unclear whether this syndrome is specific to cocaine or to impurities present in the inhaled drug (140). There is one report of cocaine-related Churg-Strauss vasculitis affecting the lung (155).
Cryptogenic Organizing Pneumonia
Cryptogenic organizing pneumonia is a rare complication of cocaine inhalation (156). Cryptogenic organizing pneumonia is an airway-centric fibrotic process with heavily muscularized bronchioles. Symptoms include dyspnea, cough, constitutional symptoms, and patchy—usually peripheral— infiltrates (140). A variety of inhalational and other insults to the lung are capable of eliciting this type of inflammatory response, but its pathogenesis is obscure (98). It is diagnosed reliably by surgical lung biopsy, and it is treated with steroids (98).
Interstitial Pulmonary Fibrosis
Pulmonary fibrosis may occur as a result of intensive or chronic use of cocaine, either inhaled or injected; silica or talc usually is found histopathologically (156). One autopsy series reported that 38% of patients had evidence of interstitial fibrosis in the lungs (157). Occasionally, the degree of fibrosis is extensive and leads to severe hypoxemia or pulmonary hypertension (156).
AMPHETAMINES AND OTHER STIMULANTS
Amphetamines increase sympathetic stimulation by causing release of biogenic amines and by inhibiting their reuptake. Effects of amphetamines are predominantly cardiovascular and neurologic (158).
Amphetamines were used in the early part of the last century to treat respiratory illness. They have sympatho-mimetic effects and can induce some bronchodilation and vasoconstriction; consequently, amphetamine inhalers were manufactured for the treatment of asthma. For rhinitis, the Benzedrine nasal inhaler introduced in 1932 contained a large dose of synthetic racemic amphetamine (134). Amphetamines are specifically retained by the lung from the circulation, a property that has been exploited for nuclear medicine imaging of the lung (159).
Amphetamines have adverse effects on the immune system, including a decrease in CD4 T-helper cells, an increase in immunosuppressive cytokines (transforming growth factor–beta and IL-10), and a switch from Th1-type cytokines (IL-2 and interferon-alpha) to Th2-type cytokines (IL-4 and IL-10) (160). Such changes may adversely affect the delayed hypersensitivity response to microbial pathogens. Chlorphentermine, an amphiphilic drug, has been reported to cause phospholipidosis in a variety of murine tissues, including the lung, which is associated with impaired phagocytosis (161).
Metabolic Acidosis and Respiratory Alkalosis
Extreme agitation and hyperthermia may result in rhabdomyolysis and severe metabolic acidosis, which will be associated with an increased respiratory drive (162). A direct central effect also may increase respiratory drive (134).
Barotrauma
Pneumomediastinum, subcutaneous emphysema, and retro-pharyngeal emphysema have been reported with the use of inhaled 3,4-methylenedioxymethamphetamine (“Ecstasy”) (134,163).
Respiratory Depression
CNS and respiratory depression may be seen, particularly in overdose. Patients are at increased risk of aspiration from a depressed mental status or from seizures (134).
Pulmonary Edema
Amphetamines may cause noncardiogenic pulmonary edema as well as myocardial infarction or acute cardio-myopathy with resulting cardiogenic pulmonary edema (134,164,165). Users also can develop pulmonary edema through a neurogenic mechanism or secondary to aspiration.
Pulmonary Hypertension
Appetite suppressants, such as fenfluramine and its derivatives, are independent risk factors for pulmonary hypertension (166). One case–control study reported that the risk of pulmonary hypertension was increased 23 times in patients who used these drugs for more than 3 months (165). The first reported association between pulmonary hypertension and amphetamine use was in Europe in the 1960s, after an increase in cases related to aminorex fumarate (167). Between 1967 and 1973, 77% of patients with pulmonary hypertension reported use of aminorex fumarate before the onset of symptoms. A 5-year retrospective study of patients referred for evaluation of pulmonary hypertension found that 15 (20%) had used fenfluramine and that 67% of these patient showed a temporal relationship between use of the drug and onset of symptoms (167). Pulmonary hypertension has been reported through chronic inhalation or through intravenous injection of methylphenidate with potentially fatal outcomes (168,169).
One possible explanation for the development of pulmonary hypertension related to fenfluramine use is that the drug increases circulating levels of serotonin and results in vasoconstriction of the pulmonary vasculature (170). Plasma serotonin levels are increased in other patients with pulmonary arterial hypertension. This excess serotonin may lead to pulmonary vasoconstriction and proliferation of pulmonary vascular smooth muscle. Other proposed mechanisms include toxic endothelial injury, hypoxia, vasospasm, vasculitis, and altered balance of mediators of vascular tone, such as eicosanoids (167,170).
Patients usually present with exertional dyspnea. On physical exam, there is a prominent second heart sound, jugular venous distension, and enlarged pulmonary vascular pedicles on chest radiograph (31). Pathology reveals changes similar to primary pulmonary hypertension, with advanced plexogenic pulmonary arteriopathy. Echocardiography can estimate pulmonary artery pressures. A right heart catheterization is necessary to confirm pulmonary pressures and to determine if pulmonary hypertension is responsive to vasodilator therapy. Vasodilator therapy with calcium channel blockers, parenteral prostanoids, or an alternative advanced agent may be used in selected individuals after hemodynamic monitoring and workup confirm the diagnosis (31).
Bullous Emphysema
Bibasilar bullous pulmonary emphysema resembling that seen in alpha-1-antitrypsin deficiency has been reported with injection of methylphenidate, but it has not been reported with amphetamines (171). The pathogenesis of this complication is not known.
CAFFEINE
Caffeine is the world’s most widely used psychoactive drug. It is widely available in food, drinks, pills, and powders. Caffeine is a phosphodiesterase inhibitor and an adenosine and benzodiazepine receptor antagonist that raises intracellular cyclic adenosine monophosphate. The effects of caffeine are similar to theophylline, including smooth muscle relaxation and mild bronchodilator properties. Caffeine may falsely elevate the serum theophylline level (172).
Pulmonary complications are rare and usually are associated with a large overdose or unintentional ingestion by children. Respiratory alkalosis, chest pain, seizures, aspiration, respiratory failure, and pulmonary edema associated with cardiac arrhythmias may occur (173).
OPIOIDS
Opioids have prominent effects on the respiratory system and other organ systems because of widespread distribution of opioid receptors throughout the body. Opioids bind to specific receptors with distribution to the CNS, cardiovascular, immune, and respiratory systems. Opioid receptors are found mostly within the alveolar walls of the respiratory tract, but they also are also present within tracheal and bronchial smooth muscle tissue (174).
Opioids have their most dramatic effects on the respiratory system by acting on the CNS. Opioid binding to μ2 receptors causes a reduction in responsiveness to carbon dioxide and depresses the pontine and medullary centers that regulate respiratory automaticity and cough (175). Cerebral cortical input also may be inhibited. Consequently, breathing becomes irregular, and apnea may develop. Respiratory depression increases progressively as the dose is increased. Maximal respiratory depression occurs within 5 to 10 minutes after an intravenous dose or within 30 to 90 minutes after a subcutaneous or intramuscular dose, with effects on respiration lasting 4 to 5 hours (175).
Opioids may induce respiratory complications indirectly through the CNS and also have effects on airways, pulmonary vasculature, and the immune system. They induce histamine release from mast cells, which may lead to pulmonary vein constriction, increased pulmonary capillary permeability and pulmonary edema, and bronchoconstriction (176). Opioids have significant effects on the immune system, which may account for the reported association with infections. Specifically, they cause defects in T-cell and natural killer cell function and macrophage and neutrophil phagocytosis. They also inhibit cytosine production by leukocytes, attenuate antibody responses, inhibit delayed-type hypersensitivity responses, decrease CD4/CD8 ratio, and inhibit leukocyte chemotaxis (177,178). Opioids can desensitize chemokine receptors in leukocytes, decreasing their chemotactic ability (179). They also can induce leukocyte apoptosis (180).
Although morphine is often used for the relief of dyspnea, opioids also exert adverse effects on the lung, acutely and with chronic abuse. Pulmonary complications account for approximately 20% of opioidrelated medical complications (181). The most common complication of opioid overdose is noncardiogenic pulmonary edema (181). Acutely, particularly with inhalational use, opioids may induce bronchospasm, bronchitis, and hypersensitivity pneumonitis. Respiratory depression and failure, pulmonary edema, respiratory infections, COPD, septic pulmonary emboli, pulmonary hypertension, and talc-related complications are associated with chronic opioid abuse (see Chapter 77 Section “Respiratory Complications of Injected Drugs”).
Bronchospasm
Heroin and opioid use can precipitate asthma exacerbations in patients with known asthma (182). Opioids cause histamine release, potentially through mu receptors or through IgE mediation, which can induce bronchospasm in histamine-sensitive asthmatics. It is unclear if histamine- insensitive asthmatics also may be susceptible to opioidinduced bronchoconstriction (183). Occupational asthma among workers in a pharmaceutical factory producing morphine was found to be associated with an IgE-mediated mechanism of histamine release (184).
Pulmonary Edema
Morphine is often used as an adjunct for the management of cardiogenic pulmonary edema. However, noncardiogenic pulmonary edema is a common complication of opioid overdose (183). This phenomenon was first described by Osler in a patient with morphine overdose. Earlier data suggested that pulmonary edema was seen in nearly 50% of patients presenting with opioid overdose; however, recent data suggest that the incidence is approximately 1% to 10% (185,186). The occurrence of pulmonary edema is not limited to intravenously administered drugs (183). Opioid-induced pulmonary edema can occur with the first use of the drug, most commonly seen in less experienced users, but it is believed to be dose-related rather than an idiosyncratic reaction (181,186).
Several mechanisms of opioid-induced noncardiogenic pulmonary edema have been proposed. One hypothesis suggests that opioids have a direct toxic effect on the alveolar–capillary membrane, increasing permeability and allowing fluid extravasation into the alveolar spaces (187). Alternatively, the opioid effect on the CNS may induce a neurogenic efferent response leading to alveolar–capillary permeability or pulmonary venous constriction (145,188). Other possibilities include a hypersensitivity reaction or acute hypoxic effect, causing increased alveolar–capillary membrane permeability (187).
Clinically, this complication manifests as dyspnea and somnolence, usually within minutes, depending on the route of administration. Bilateral crackles may be heard on physical examination. Affected individuals become progressively hypoxemic and hypercapnic, leading to cyano-sis and obtundation (187). The chest radiograph typically reveals interstitial and/or alveolar bilateral infiltrates, often in a perihilar pattern, without cardiomegaly or pleural effusions (183). The pulmonary capillary wedge pressure usually is within the normal range. The electrocardiogram may be normal or may show arrhythmias and conduction defects (183).
Treatment is supportive and may include noninvasive or invasive mechanical ventilation, oxygen, and judicious use of diuretics (183). The clinical and radiographic abnormalities generally clear within 24 hours. If there is no improvement after 48 hours, alternative diagnoses should be considered, including aspiration and superimposed pneumonia, which may affect at least 50% opioid abusers with pulmonary edema (183).
Sleep-Disordered Breathing
Chronic opioid use is associated with increased rates of central and obstructive sleep apnea. In one observational study, 75% of these patients had sleep apnea (189). Methadone in particular is associated with increased sleep-disordered breathing with a relationship between methadone dose and duration of use and the apnea–hypopnea index (190,191). Opioid use has also been associated with decreased effectiveness of continuous positive airway pressure therapy for obstructive sleep apnea (192).
Hypersensitivity Pneumonitis
Although uncommon, intranasal heroin has been reported to cause hypersensitivity pneumonitis (193). Patients usually manifest with dyspnea and cough hours to days after inhalation, which may lead to significant hypoxemia. Chest radiograph may show bilateral infiltrates, which may be reticulonodular or coalescent. Treatment includes supplemental oxygen and steroids if significant gas exchange compromise is evident or if spontaneous regression does not occur (193).
ALCOHOL
Alcohol ingestion may cause acute intoxication accompanied by respiratory depression and common pulmonary complications including atelectasis, hypoxemia, respiratory failure, respiratory acidosis, aspiration, and ARDS (194). Aspiration pneumonia may be associated with loss of airway control or seizures. Alcohol ingestion also may cause worsening of underlying respiratory conditions such as sleep-disordered breathing (195). Alcohol depresses respiration by acting directly on the respiratory centers within the brain. Chronic heavy use may lead to cirrhosis, with the development of specific cirrhosis-related pulmonary syndromes and increased susceptibility to infections. Pneumonia related to Klebsiella, Streptococcus pneumoniae, Acinetobacter, tuberculosis, and other pathogens is common among individuals with recurrent heavy drinking (196,197). This condition may stem from malnutrition, concomitant tobacco use, immunosuppression, and decreased function of the reticu-loendothelial system (198,199).
Acute Metabolic Acidosis and Respiratory Alkalosis
Alcohol may cause metabolic acidosis from alcoholic keto-acidosis, with resultant compensatory respiratory alkalosis (200). The consumption of other toxic alcohols, including isopropanol, ethylene glycol, and methanol, continues to be a public health issue. Patients presenting with alcohol intoxication and high anion gap metabolic acidosis should be evaluated for the presence of an unexplained osmolar gap, which increases the concern for a coingestion (201,202).
Chronic Respiratory Alkalosis
Among patients with cirrhosis, chronic respiratory alkalo-sis is common, even in the absence of metabolic acidosis (203). This condition may be due to the respiratory stimulant effect of poorly cleared progesterone and estradiol on the CNS (203).
Asthma
Patients with asthma may experience worsening of asthma symptoms after consumption of alcohol, particularly those who are histamine sensitive (204). Additionally, alcohol use is associated with adult-onset asthma (205). In a Northern European survey, 10% of alcohol users experienced alcohol-induced airway symptoms (204). In some individuals, alcohol consumption can result in bronchodilation; this may be due to the presence of sulfites in wine or other beverages (206). In a large observational study, low amounts of alcohol use were associated with higher FEV1, FVC, and FEV1/FVC than no use (207). However, airflow obstruction increases in former heavy drinkers (207). In some patients, acetaldehyde, generated from ethanol metabolism, leads to mast cell or basophil degranulation (208). The ensuing release of histamine and other mediators of inflammation induces bronchospasm, as suggested by the ability of chlorpheniramine largely to inhibit ethanol-induced bronchospasm (209). Additionally, alcohol increases the total level of serum IgE and impairs T-helper lymphocyte type 1 immune responses (205). Both heterozygotic and homozygotic mutations in acetaldehyde dehydrogenase genes have been found to correlate with alcohol-induced airway reactivity (210).
Hepatic Hydrothorax
Cirrhosis with ascites may lead to pleural effusions (211). Negative intrapleural pressure generated during inspiration leads to transdiaphragmatic movement of ascitic fluid into the pleural space. Ascitic fluid often occupies much of the hemi-thorax, usually on the right side (211). If refractory to diuretics and other medical therapy, pleurodesis, pleural drainage catheters, or transjugular intrahepatic portosystemic shunts may be performed with variable success (212–214).
Pulmonary Restriction from Ascites
Massive ascites, with or without hepatic hydrothorax, may restrict diaphragmatic and pulmonary excursion, leading to rapid shallow breathing, dyspnea, atelectasis, and hypoxemia in severe cases (215,216).
Hepatopulmonary Syndrome
Hepatopulmonary syndrome (HPS) affects 15% to 30% of patients with cirrhosis. HPS consists of portal hypertension, intrapulmonary microvascular vasodilation, and an increased alveolar–arterial oxygen gradient with hypoxemia in patients with cirrhosis (217). Vasodilations in the pre– and post–capillary pulmonary vasculature are associated with increased pulmonary nitric oxide production, increased macrophage production of heme oxygenase 1, and increased activation of angiogenic pathways by monocytes.
Symptoms include dyspnea, orthodeoxia and platypnea, clubbing, and cyanosis. The classic symptom of orthodeoxia and finding of platypnea are attributed to positional increase in vasodilation and shunting through the lower lobes. Diagnostic elements include sitting and supine pulse oximetry or arterial blood gas measurements and contrast echocardiography with agitated saline to identify intrapul-monary shunts. Alternative imaging techniques, including radionucleotide imaging with a technetium 99-macroaggre-gated albumin perfusion scan, high-resolution chest computed tomography, and pulmonary angiography, are less sensitive than contrast echocardiography. Liver transplant reverses HPS. There is inconclusive evidence for the use of transjugular intrahepatic portosystemic shunts and pent-oxifylline (217).
Portopulmonary Hypertension
Pulmonary arterial hypertension affects approximately 2% to 5% of patients with cirrhosis and portal hypertension (217). The pathogenesis of this complication is not clear, but the development has been associated with a number of vasoactive substances including endothelin 1, prostacyclin, and throm-boxane; genetic risk factors; shear stress; and inflammatory processes. These mediators result in a number of changes in the pulmonary arterial vessels, including smooth muscle hyperplasia and hypertrophy, plexogenic pulmonary arteriopathy, and cellular proliferation and fibrosis in the intima.
Patients usually have exertional dyspnea with possible fatigue, orthopnea, chest pain, or syncope. The physical signs of pulmonary hypertension include prominent P2 and tricuspid regurgitation murmur; other findings such as ascites and peripheral edema are difficult to assess in cirrhotic patients who may have these findings secondary to their portal hypertension. Initial diagnostic testing is performed with transthoracic echocardiography. If there is evidence of increased right ventricular systolic pressure, then a confirmatory right heart catheterization should be performed. Patients with portopulmonary hypertension should be referred to a specialized pulmonary hypertension center for evaluation of advanced pulmonary artery vasodilator therapies. Liver transplant as a primary treatment remains controversial (217). A select group of patients have better outcomes with a combination of medical therapy and liver transplant; however, transplantation in fixed pulmonary hypertension portends a poor prognosis (217,218). Transjugular intrahepatic portosystemic shunts should not be performed to treat portopulmonary hypertension (219).
Acute Respiratory Distress Syndrome
Individuals who drink heavily or have alcohol use disorders are twice as likely to develop ARDS and to die from ARDS once it develops. Heavy drinkers and those with alcohol use disorders with sepsis are more likely to develop ARDS and are at increased risk for transfusion-related acute lung injury (198). Epithelial lining fluid from the lungs of heavy alcohol users is depleted of glutathione, which is important in mitigating the oxidative stress that plays a role in the pathogenesis of ARDS (220). Additionally, patients with alcohol use disorders have impaired immune function, decreased alveolar epithelial cell function, altered activation of the renin– angiotensin system, and impaired granulocyte–macrophage colony-stimulating factor activity, all of which are thought to play a role in the pathogenesis of ARDS (198).
SEDATIVE–HYPNOTICS
Sedative–hypnotic drugs may exert significant respiratory depressant effects when abused or when mixed with alcohol and opiates. Benzodiazepines, barbiturates, gamma-hydroxybutyrate (GHB), and zolpidem bind gamma-aminobutyric acid (GABA) receptors, which normally function as targets for inhibitory neurotransmitters and promote sedation, hypnosis, anxiolysis, anterograde amnesia, and anticonvulsant activity (175).
GHB is a naturally occurring metabolite of GABA in the CNS (221). Unlike benzodiazepines and barbiturates that bind GABA-A receptors, GHB interacts with GABA-B receptors. Other GHB effects include elevation of CNS dopamine, elevation of CNS endorphins, and stimulation of growth hormone release. GHB is abused by bodybuilders and by those seeking its hypnotic and euphoric effects (222). Adverse respiratory effects are mainly related to respiratory depression from overdose.
Overdose
Barbiturates were the cornerstone of sedative–hypnotic therapy until the 1970s, when they were replaced by less toxic benzodiazepines. Data compiled from the U.S. Poison Control Centers found sedatives and hypnotics to be the fourth most frequent category of substance-related adverse events, and they are the leading cause of death related to substance exposure (223). Benzodiazepine and pharmaceutical opioids are the most common cause of mortality from polysubstance overdose (224).
Although barbiturates also may be involved in a poly-pharmacy overdose, the most common toxic scenario with barbiturates is an accidental or intentional oral ingestion by a seizure patient or member of his or her family (225). Barbiturate overdose is more likely to result in coma or death secondary to increased risk of respiratory depression compared to benzodiazepines (226). Zolpidem, a nonben-zodiazepine hypnotic, has been reported to cause respiratory failure and coma with acute overdose (227).
Clinically, sedation may progress to coma, with progressive alveolar hypoventilation and respiratory acidosis. Patients are at increased risk for aspiration and atelectasis. Hypotension, which probably is related to direct myocardial depression and vasodilation, may follow, with accompanying respiratory arrest (more typically in barbiturate overdose) (225).
Treatment of sedative–hypnotic overdose is supportive. Decontamination with lavage and charcoal adsorption may be appropriate in patients who present within a narrow window (1 hour) from ingestion. Supplemental oxygen, airway protection, and mechanical ventilation may be necessary. Most deaths occur as a result of ARDS secondary to either a chemical aspiration pneumonitis or bacterial pneumonia (175).
In benzodiazepine overdose, the competitive antagonist flumazenil can be administered with caution, but it is short acting, and its ability to reverse the respiratory depressant effects is controversial (226). Side effects include anxiety, agitation, crying, and nausea (225). Seizures can be precipitated in patients with physiologic dependence (226). In barbiturate overdose, elimination can be enhanced with an alkaline diuresis (226). Dialysis rarely is necessary; however, hemodynamic instability refractory to fluid management is an indication for dialysis (228). Serum drug levels often rebound after dialysis because of redistribution, necessitating further treatment (228).
The use of CNS stimulants may increase mortality and is contraindicated (175,226). In GHB overdose, recovery is rapid, with full return to baseline within several hours (221). Recovery from benzodiazepine and barbiturate overdose is longer in duration (226). Generally, the prognosis is excellent with supportive care alone.
Withdrawal Syndromes
Abrupt cessation of chronically used sedative–hypnotics may lead to withdrawal symptoms (225). Benzodiazepine withdrawal causes a hyperadrenergic state that increases carbon dioxide production. Tachypnea is the most common respiratory manifestation of withdrawal from benzodiaz-epines. Tachypnea is accompanied by other manifestations of benzodiazepine withdrawal, including anxiety, tremor, headache, diaphoresis, difficulty concentrating, insomnia, hallucinations, fatigue, tachycardia, hypertension, and seizure. Increased minute ventilation is seen due to increased carbon dioxide production and anxiety in patients experiencing withdrawal. It generally occurs 2 to 5 days after the last dose of the drug (225). Management typically requires a gradual taper of benzodiazepine (229). Additionally, car-bamazepine and valproate have a beneficial role, and there is increasing evidence for the role of gabapentin, pregabalin, and topiramate (229).
Barbiturate withdrawal occurs after 2 to 7 days of abstinence (225). Agitation, hyperreflexia, anxiety, and tremor are most common, followed by confusion and hallucinations. Up to 75% of patients experience seizures, often refractory to phenytoin. Effective treatment requires reinstitution of a barbiturate or a cross-tolerant medication such as a benzodiazepine. Because seizures are so common in barbiturate withdrawal, airway protection and mechanical ventilation management are the primary respiratory issues (225).
After prolonged abuse of GHB, physiologic dependence can occur (230). Initial withdrawal symptoms are similar to those in alcohol or benzodiazepine withdrawal. Later, cho-reatic movements and restlessness of the tongue develop. Loss of airway protective reflexes may necessitate airway protection and mechanical ventilation (230). Zolpidem withdrawal is rarely reported. In patients taking 20 to 30 times the recommended dose over long periods of time, withdrawal has been characterized as restlessness, diaphoresis, tremors, and seizures resulting in respiratory failure and aspiration (231).
Sleep-Disordered Breathing
Benzodiazepines can worsen sleep-disordered breathing by decreasing the tone of the upper airway muscles resulting in increased obstructive events. Increased central apneas and reduced ventilatory response to carbon dioxide also lead to worse nocturnal hypoxemia (232). Although benzodiazepines can be beneficial in treating sleep disorders by increasing total sleep time and the sense of refreshing sleep, they result in a loss of stage 3 deep sleep and overall less restorative sleep (232). Zolpidem improves sleep fragmentation and does not decrease time in deep sleep. However, it results in increased obstructive apneic events and decreased oxygen saturation during each event (233). GHB does not increase obstructive events, but it may increase central apneas and cause increased nocturnal desaturation in some patients (234).
VOLATILE SUBSTANCES
Abuse of inhalants is highly prevalent throughout the world, particularly among young teenagers; however, the prevalence of reports to poison control centers has decreased 33% from 1993 to 2008 (235). Volatile substances are aromatic and short-chain hydrocarbons, such as toluene, gasoline, butane, butyl and amyl nitrites, and organofluorines that are found in adhesives, paints, paint thinners, dry cleaning fluids, refrigerants, and propellants (236). They are volatile at room temperature. When sniffed or vigorously inhaled within a hermetic container (“huffed”), they are readily absorbed in the lungs. Intoxicating and dysphoric effects follow within seconds. Primary acute effects involve the CNS and include lethargy, stupor, agitation, hallucinations, dizziness, and seizures (236,237). Pulmonary complications include severe respiratory depression, barotrauma (pneumomediastinum), persistent cough, noncardiogenic pulmonary edema, and asphyxiation (66,237,238).
Methemoglobinemia
Butyl and isobutyl nitrites may cause methemoglobinemia, which manifests as cyanosis with low oxygen saturation of hemoglobin but with normal partial pressure of oxygen (238). CO-oximetry will specifically measure methemoglobin levels (239). Intravenous methylene blue may be administered for treatment (239).
Metabolic Acidosis and Respiratory Alkalosis
Metabolic acidosis may occur, with compensatory respiratory alkalosis, resulting from distal renal tubular acidosis or from increased anion gap acidosis (240). The latter may result from oxidation of toluenes to benzoic acid and conjugation with glycine to form hippuric acid (240).
Asthma
Occupational exposure to toluene diisocyanate and other isocyanates is associated with asthma (241). However, no cases of asthma associated with toluene abuse have been reported. In some cases, an IgE-mediated mechanism may be operative, but in others, multiple mechanisms are responsible (242). A hypersensitivity pneumonitis also has been reported with isocyanate inhalation (243).
Tracheobronchitis
Nitrates and other inhalants may be directly irritating to airway mucosa, which can cause chronic cough (244).
Asphyxiation
Users of inhalants may develop asphyxiation from plastic bag suffocation, respiratory depression, displacement of oxygen in the inspired air, and acute noncardiogenic pulmonary edema (66,67).
NITROUS OXIDE
Nitrous oxide is a readily available and often misused substance with euphoric effects. It is a widely used inha-lational anesthetic–analgesic agent that also is used in a variety of commercial products (such as a propellant for whipped cream chargers) (245). Pulmonary complications include pneumomediastinum, respiratory depression, and hypoxemia because of displacement of oxygen, leading to asphyxiation (245–247). Treatment is supportive, including supplemental oxygen and respiratory support (245).
ANABOLIC STEROIDS
Anabolic steroids induce a prothrombotic state and may cause pulmonary embolism, strokes, and other forms of thrombosis (248–250). Other respiratory complications are less common but may occur after stroke and include atelectasis, pneumonia, aspiration, neurogenic pulmonary edema, and sleep-disordered breathing (251).
REFERENCES
1.West J. Respiratory physiology: the essentials. 8th ed. Baltimore, MD: Lippincott Williams & Wilkins, 2008.
2.Brugman S, Irvin C. Lung structure and function. In: RS Mitchell, T Petty, M Schwartz, eds. Synopsis of clinical pulmonary disease, 4th ed. St. Louis, MO: Mosby, 1989:1–17.
3.Wilson KC, Saukkonen JJ. Acute respiratory failure from abused substances. J Intensive Care Med 2004;19(4):183–193.
4.Hedenstierna G, Edmark L. Mechanisms of atelectasis in the perioperative period. Best Pract Res Clin Anaesthesiol 2010;24(2):157–169.
5.Chen A, Kollef MH. Bronchoscopy in the intensive care unit. In: K Wang, A Mehta, JF Turner, eds. Flexible bronchoscopy, 3rd ed. Hoboken, NJ: Wiley-Blackwell, 2011:317.
6.Adnet F, Baud F. Relation between Glasgow Coma Scale and aspiration pneumonia. Lancet 1996;348(9020):123–124.
7.Marik PE. Pulmonary aspiration syndromes. Curr Opin Pulm Med 2011;17(3):148–154.
8.Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 2001;344(9):665–671.
9.Burnham EL, Phang TL, House R, et al. Alveolar macrophage gene expression is altered in the setting of alcohol use disorders. Alcohol Clin Exp Res 2011;35(2):284–294.
10.Kaushik KS, Kapila K, Praharaj AK. Shooting up: the interface of microbial infections and drug abuse. J Med Microbiol 2011;60(Pt 4): 408–422.
11.McPhee SJ, Papadakis MA, Rabow MW. Current medical diagnosis and treatment 2012, 51st ed. New York, NY: McGraw-Hill Medical, 2012.
12.Cole C, Jones L, McVeigh J, et al. Adulterants in illicit drugs: a review of empirical evidence. Drug Test Anal 2011;3(2):89–96.
13.Chang A, Osterloh J, Thomas J. Levamisole: a dangerous new cocaine adulterant. Clin Pharmacol Ther 2010;88(3): 408–411.
14.Kagen SL, Kurup VP, Sohnle PG, et al. Marijuana smoking and fungal sensitization. J Allergy Clin Immunol 1983;71(4):389–393.
15.Bal A, Agarwal AN, Das A, et al. Chronic necrotising pulmonary aspergillosis in a marijuana addict: a new cause of amyloidosis. Pathology 2010;42(2):197–200.
16.Trullas J-C, Bisbe J, Miro JM. Aspergillosis in drug addicts. In: AC Pasqualotto, ed.Aspergillosis: from diagnosis to prevention. New York, NY: Springer, 2010:245–558.
17.Cha ES, Lee YK, Moon EK, et al. Paraquat application and respiratory health effects among South Korean farmers. Occup Environ Med 2012;69(6):398–403.
18.Beligaswatte AM, Kularatne SA, Seneviratne AB, et al. An outbreak of fatal pneumonitis caused by contamination of illicit alcohol with paraquat. Clin Toxicol (Phila) 2008;46(8):768–770.
19.Da Silva FR, Da Silva J, Allgayer Mda C, et al. Genotoxic biomonitoring of tobacco farmers: biomarkers of exposure, of early biological effects and of susceptibility. J Hazard Mater2012;225-226(0):81–90.
20.Kim J, Ko Y, Lee WJ. Depressive symptoms and severity of acute occupational pesticide poisoning among male farmers. Occup Environ Med 2013;70(5):303–309.
21.Han B, Gfroerer JC, Colliver JD. Associations between duration of illicit drug use and health conditions: results from the 2005–2007 national surveys on drug use and health. Ann Epidemiol 2010;20(4):289–297.
22.Story A, Bothamley G, Hayward A. Crack cocaine and infectious tuberculosis. Emerg Infect Dis 2008;14(9):1466–1469.
23.Maldonado F, Limper AH. Drug-induced lung diseases. Interstitial Lung Disease. 2011:280.
24.Marchiori E, Lourenco S, Gasparetto TD, et al. Pulmonary talcosis: imaging findings. Lung 2010;188(2):165–171.
25.Overland ES, Nolan AJ, Hopewell PC. Alteration of pulmonary function in intravenous drug abusers. Prevalence, severity, and characterization of gas exchange abnormalities. Am J Med1980;68(2):231–237.
26.Pare JP, Cote G, Fraser RS. Long-term follow-up of drug abusers with intravenous talcosis. Am Rev Respir Dis 1989;139(1):233–241.
27.Griffith CC, Raval JS, Nichols L. Intravascular talcosis due to intravenous drug use is an underrecognized cause of pulmonary hypertension. Pulm Med 2012;2012:617531.
28.Price L, Bouillon K, Wort SJ, et al. Drug- and toxin-induced pulmonary arterial hypertension. In: M Humbert, R Souza, G Simonneau, eds. Pulmonary vascular disorders. Basel, Karger. 2012;41:76–84.
29.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54(1 Suppl):S43–S54.
30.Collazos J, Martinez E, Fernandez A, et al. Acute, reversible pulmonary hypertension associated with cocaine use. Respir Med 1996;90(3):171–174.
31.Nef HM, Mollmann H, Hamm C, et al. Pulmonary hypertension: updated classification and management of pulmonary hypertension. Heart 2010;96(7):552–559.
32.McLean S, Bruno R, Brandon S, et al. Effect of filtration on morphine and particle content of injections prepared from slow-release oral morphine tablets. Harm Reduct J 2009;6:37.
33.Patel P, Patel RP, Brandon S, et al. Effects of filtration on the presence of particulate and oxycodone content of injections prepared from crushed OxyContin(R) tablets. Curr Drug Saf2012;7(3):218–224.
34.Roux P, Carrieri MP, Keijzer L, et al. Reducing harm from injecting pharmaceutical tablet or capsule material by injecting drug users. Drug Alcohol Rev 2011;30(3):287–290.
35.Gordon RJ, Lowy FD. Bacterial infections in drug users. N Engl J Med 2005;353(18):1945–1954.
36.Lloyd-Smith E, Wood E, Zhang R, et al. Determinants of hospitalization for a cutaneous injection-related infection among injection drug users: a cohort study. BMC Public Health2010;10(1):327.
37.Saydain G, Singh J, Dalal B, et al. Outcome of patients with injection drug use-associated endocarditis admitted to an intensive care unit.J Crit Care 2010;25(2):248–253.
38.Poole PS. Necrotizing pneumonia, pulmonary abscess, and lung gangrene: multiple pathways to lung parenchymal destruction. Contemp Diagn Radiol 2011;34(13):1.
39.Yazbeck MF, Dahdel M, Kalra A, et al. Lung abscess: update on microbiology and management. Am J Ther 2012;1536–86.
40.Monroe EJ, Tailor TD, McNeeley MF, et al. Needle embolism in intravenous drug abuse. Radiol Case Rep 2012;7(3):1–4.
41.Galante JM, Ahmad S, Albers EA, et al. Trauma and substance abuse: deadly consequences of intravenous percocet tablets. J Emerg Med 2012;43(3):e167–e169.
42.Zorc TG, O’Donnell AE, Holt RW, et al. Bilateral pyopneumothorax secondary to intravenous drug abuse. Chest 1988;93(3):645–647.
43.Haynes D, Baumann MH. Management of pneumothorax. Semin Respir Crit Care Med 2011;31(6):769–780.
44.Müller KAL, Zürn CS, Patrik H, et al. Unusual association of diseases/symptoms: massive haemoptysis in an intravenous drug user with infective tricuspid valve endocarditis. BMJ Case Rep2010;2010:bcr1020092404.
45.McLean L, Sharma S, Maycher B. Mycotic pulmonary arterial aneurysms in an intravenous drug user. Can Respir J 1998;5(4):307–311.
46.Whayne NG, Spitz WU. Rupture of a subclavian artery aneurysm in a heroin addict. Report of a case. Z Rechtsmed 1978;81(2):147–149.
47.Stein MD. Medical complications of intravenous drug-use. J Gen Intern Med 1990;5(3):249–257.
48.Cone EJ. Recent discoveries in pharmacokinetics of drugs of abuse. Toxicol Lett 1998;102–103:97–101.
49.Sandford AJ, Chagani T, Weir TD, et al. Susceptibility genes for rapid decline of lung function in the lung health study. Am J Respir Crit Care Med 2001;163(2):469–473.
50.Hakim F, Hellou E, Goldbart A, et al. The acute effects of water-pipe smoking on the cardiorespiratory system. Chest 2011;139(4):775–781.
51.Raad D, Gaddam S, Schunemann HJ, et al. Effects of water-pipe smoking on lung function: a systematic review and meta-analysis. Chest 2011;139(4):764–774.
52.Smith CJ, Hansch C. The relative toxicity of compounds in mainstream cigarette smoke condensate. Food Chem Toxicol 2000;38(7):637–646.
53.Palmer F, Jaffray M, Moffat MA, et al. Prevalence of common chronic respiratory diseases in drug misusers: a cohort study. Prim Care Respir J 2012;21(4):377–383.
54.Johnson MK, Smith RP, Morrison D, et al. Large lung bullae in marijuana smokers. Thorax 2000;55(4):340–342.
55.Beshay M, Kaiser H, Niedhart D, et al. Emphysema and secondary pneumothorax in young adults smoking cannabis. Eur J Cardiothorac Surg 2007;32(6):834–838.
56.Rome LA, Lippmann ML, Dalsey WC, et al. Prevalence of cocaine use and its impact on asthma exacerbation in an urban population. Chest 2000;117(5):1324–1329.
57.Tashkin DP. Airway effects of marijuana, cocaine, and other inhaled illicit agents. Curr Opin Pulm Med 2001;7(2):43–61.
58.Osborn HH, Tang M, Bradley K, et al. New-onset bronchospasm or recrudescence of asthma associated with cocaine abuse. Acad Emerg Med 1997;4(7):689–692.
59.Venkatanarasimha N, Rock B, Riordan RD, et al. Imaging of illicit drug use. Clin Radiol 2010;65(12):1021–1030.
60.Hazouard E, Koninck JC, Attucci S, et al. Pneumorachis and pneumomediastinum caused by repeated Muller’s maneuvers: complications of marijuana smoking. Ann Emerg Med2001;38(6):694–697.
61.Lanteri A, Bhangle SD, Fless KG. Marijuana use and the development of status asthmaticus: a case report. OJAnes 2012;2(3):62–64.
62.Seaman ME. Barotrauma related to inhalational drug abuse. J Emerg Med 1990;8(2):141–149.
63.McCarroll KA, Roszler MH. Lung disorders due to drug abuse. J Thorac Imaging 1991;6(1):30–35.
64.Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol 2011;30(10):1385–1392.
65.Karila L, Petit A, Lowenstein W, et al. Diagnosis and consequences of cocaine addiction. Curr Med Chem 2012;19(33):5612–5618.
66.Vance C, Swalwell C, McIntyre I. Deaths involving 1,1-difluoroethane at the San Diego County Medical Examiner’s Office. J Anal Toxicol 2012;36(9):626–633.
67.Sakai K, Maruyama-Maebashi K, Takatsu A, et al. Sudden death involving inhalation of 1,1-difluoroethane (HFC-152a) with spray cleaner: three case reports. Forensic Sci Int2011;206(1–3):e58–e61.
68.Ishii T, Matsuse T, Teramoto S, et al. Association between alpha-1-antichymotrypsin polymorphism and susceptibility to chronic obstructive pulmonary disease. Eur J Clin Invest2000;30(6):543–548.
69.Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol 2009;9(5):377–384.
70.Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med2013;187(4):347–365.
71.Lee PN, Fry JS. Systematic review of the evidence relating FEV1 decline to giving up smoking. BMC Med 2010;8(1):84.
72.Vassallo R, Ryu JH. Tobacco smoke-related diffuse lung diseases. Semin Respir Crit Care Med 2008;29(6):643–650.
73.Caminati A, Cavazza A, Sverzellati N, et al. An integrated approach in the diagnosis of smoking-related interstitial lung diseases. Eur Respir Rev 2012;21(125):207–217.
74.Oberg M, Jaakkola MS, Woodward A, et al. Worldwide burden of disease from exposure to second-hand smoke: a retrospective analysis of data from 192 countries. Lancet2011;377(9760):139–146.
75.Tinuoye O, Pell JP, Mackay DF. Meta-analysis of the association between secondhand smoke exposure and physician-diagnosed childhood asthma. Nicotine Tob Res 2013;15(9):1475–1483.
76.Oono IP, Mackay DF, Pell JP. Meta-analysis of the association between secondhand smoke exposure and stroke. J Public Health (Oxf) 2011;33(4):496–502.
77.Taylor R, Najafi F, Dobson A. Meta-analysis of studies of passive smoking and lung cancer: effects of study type and continent. Int J Epidemiol 2007;36(5):1048–1059.
78.Balfour D, Benowitz N, Fagerstrom K, et al. Diagnosis and treatment of nicotine dependence with emphasis on nicotine replacement therapy. A status report. Eur Heart J2000;21(6):438–445.
79.Berlin I. Therapeutic strategies to optimize the efficacy of nicotine replacement therapies. COPD 2009;6(4):272–276.
80.Islam N, Rahman S. Improved treatment of nicotine addiction and emerging pulmonary drug delivery. Drug Discov Ther 2012;6(3):123–132.
81.Caldwell B, Dickson S, Burgess C, et al. A pilot study of nicotine delivery to smokers from a metered-dose inhaler. Nicotine Tob Res 2009;11(4):342–347.
82.Bullen C, McRobbie H, Thornley S, et al. Effect of an electronic nicotine delivery device (e cigarette) on desire to smoke and withdrawal, user preferences and nicotine delivery: randomised cross-over trial. Tob Control 2010;19(2):98–103.
83.Caponnetto P, Campagna D, Papale G, et al. The emerging phenomenon of electronic cigarettes. Exp Rev Respir Med 2012;6(1):63–74.
84.Lundback B, Lindberg A, Lindstrom M, et al. Not 15 but 50% of smokers develop COPD?—Report from the Obstructive Lung Disease in Northern Sweden Studies. Respir Med2003;97(2):115–122.
85.Hoyert DL, Arias E, Smith BL, et al. National vital statistics reports. Deaths: Final Data for 2012. 2012;61.
86.Vaz Fragoso CA, Concato J, McAvay G, et al. The ratio of FEV1 to FVC as a basis for establishing chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010;181(5):446–451.
87.Verra F, Escudier E, Lebargy F, et al. Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am J Respir Crit Care Med 1995;151(3 Pt 1):630–634.
88.Polkey MI, Hamnegard CH, Hughes PD, et al. Influence of acute lung volume change on contractile properties of human diaphragm. J Appl Physiol 1998;85(4):1322–1328.
89.Casale TB, Rhodes BJ, Donnelly AL, et al. Airway responses to methacholine in asymptomatic nonatopic cigarette smokers. J Appl Physiol 1987;62(5):1888–1892.
90.Hahn HL, Lang M, Bleicher S, et al. Nicotine-induced airway smooth muscle contraction: neural mechanisms involving the airway epithelium. Functional and histologic studies in vitro. Clin Investig1992;70(3–4):252–262.
91.Hong JL, Rodger IW, Lee LY. Cigarette smoke-induced bronchoconstriction: cholinergic mechanisms, tachykinins, and cyclooxygenase products. J Appl Physiol 1995;78(6): 2260–2266.
92.Hong JL, Lee LY. Cigarette smoke-induced bronchoconstriction: causative agents and role of thromboxane receptors. J Appl Physiol 1996;81(5):2053–2059.
93.Turato G, Zuin R, Saetta M. Pathogenesis and pathology of COPD. Respiration 2001;68(2):117–128.
94.Finkelstein R, Fraser RS, Ghezzo H, et al. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med 1995;152(5 Pt 1):1666–1672.
95.Health NIo. National Asthma Education and Prevention Program. Full Report of the Expert Panel: Guidelines for the diagnosis and management of asthma (EPR-3) 2007. 2011.
96.Vassallo R, Ryu JH. Smoking-related interstitial lung diseases. Clin Chest Med 2012;33(1):165–178.
97.Donaghy M, Rees AJ. Cigarette smoking and lung haemorrhage in glomerulonephritis caused by autoantibodies to glomerular basement membrane. Lancet 1983;2(8364):1390–1393.
98.Travis W, King T, Bateman E, et al. ATS/ERS International multidisciplinary consensus classification of idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2002;165(2):277–304.
99.Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J 2008;32(5):1371–1385.
100.Cheng YL, Huang TW, Lin CK, et al. The impact of smoking in primary spontaneous pneumothorax. J Thorac Cardiovasc Surg 2009;138(1):192–195.
101.Milroy CM, Parai JL. The histopathology of drugs of abuse. Histopathology 2011;59(4):579–593.
102.Cruz D, Tanoue LT, Matthay RA. Lung cancer: epidemiology, etiology, and prevention. ClinChest Med 2011;32(4):605–644.
103.National Lung Screening Trial Research Team. Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med2011;365(5):395–409.
104.Hurt RD, Ebbert JO, Hays JT, et al. Preventing lung cancer by treating tobacco dependence. Clin Chest Med 2011;32(4): 645–657.
105.Godtfredsen NS, Prescott E, Osler M. Effect of smoking reduction on lung cancer risk. JAMA 2005;294(12):1505–1510.
106.Huuskonen MS, Husman K, Jarvisalo J, et al. Extrinsic allergic alveolitis in the tobacco industry. Br J Ind Med 1984;41(1):77–83.
107.Lander F, Jepsen JR, Gravesen S. Allergic alveolitis and late asthmatic reaction due to molds in the tobacco industry. Allergy 1988;43(1):74–76.
108.Pauly JL, Smith LA, Rickert MH, et al. Review: is lung inflammation associated with microbes and microbial toxins in cigarette tobacco smoke? Immunol Res 2010;46(1–3):127–136.
109.Miller GJ, Ashcroft MT, Beadnell HM, et al. The lipoid pneumonia of blackfat tobacco smokers in Guyana. Q J Med 1971;40(160):457–470.
110.Marchiori E, Zanetti G, Mano CM, et al. Exogenous lipoid pneumonia. Clinical and radiological manifestations. Respir Med 2011;105(5):659–666.
111.King BA, Alam S, Promoff G, et al. Awareness and ever use of electronic cigarettes among U.S. adults, 2010–2011. Nicotine Tob Res 2013;15(9):1623–7.
112.FDA. Summary of results: laboratory analysis of electronic cigarettes conducted by FDA. US Department of Health and Human Services website; 2009.
113.Vardavas CI, Anagnostopoulos N, Kougias M, et al. Short-term pulmonary effects of using an electronic cigarette impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest2012;141(6):1400–1406.
114.Van Hoozen BE, Cross CE. Marijuana. Respiratory tract effects. Clin Rev Allergy Immunol. 1997;15(3):243–269.
115.Wu TC, Tashkin DP, Djahed B, et al. Pulmonary hazards of smoking marijuana as compared with tobacco. N Engl J Med 1988;318(6):347–351.
116.Baldwin GC, Tashkin DP, Buckley DM, et al. Marijuana and cocaine impair alveolar macrophage function and cytokine production. Am J Respir Crit Care Med 1997;156(5):1606–1613.
117.Sarafian TA, Magallanes JA, Shau H, et al. Oxidative stress produced by marijuana smoke. An adverse effect enhanced by cannabinoids. Am J Respir Cell Mol Biol 1999;20(6):1286–1293.
118.Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007;62(12):1058–1063.
119.Zhang ZF, Morgenstern H, Spitz MR, et al. Marijuana use and increased risk of squamous cell carcinoma of the head and neck. Cancer Epidemiol Biomarkers Prev 1999;8(12):1071–1078.
120.Pletcher MJ, Vittinghoff E, Kalhan R, et al. Association between marijuana exposure and pulmonary function over 20 years. JAMA 2012;307(2):173–181.
121.Tetrault JM, Crothers K, Moore BA, et al. Effects of marijuana smoking on pulmonary function and respiratory complications: a systematic review. Arch Intern Med 2007;167(3):221–228.
122.Vethanayagam D, Pugsley S, Dunn EJ, et al. Exogenous lipid pneumonia related to smoking weed oil following cadaveric renal transplantation. Can Respir J 2000;7(4):338–342.
123.Lai PS, Christiani DC. Long-term respiratory health effects in textile workers. Curr Opin Pulm Med 2013;19(2):152–157.
124.Kreek MJ. Cocaine, dopamine and the endogenous opioid system. J Addict Dis 1996;15(4):73–96.
125.Fattinger K, Benowitz NL, Jones RT, et al. Nasal mucosal versus gastrointestinal absorption of nasally administered cocaine. Eur J Clin Pharmacol 2000;56(4):305–310.
126.McBride DC, Inciardi JA, Chitwood DD, et al. Crack use and correlates of use in a national population of street heroin users. The National AIDS Research Consortium. J Psychoactive Drugs1992;24(4):411–416.
127.Hatsukami DK, Fischman MW. Crack cocaine and cocaine hydrochloride. Are the differences myth or reality? JAMA 1996;276(19):1580–1588.
128.Tashkin DP, Kleerup EC, Hoh CK, et al. Effects of ‘crack’ cocaine on pulmonary alveolar permeability. Chest 1997;112(2):327–335.
129.Tashkin DP, Gorelick D, Khalsa ME, et al. Respiratory effects of cocaine freebasing among habitual cocaine users. J Addict Dis 1992;11(4):59–70.
130.Baldwin GC, Roth MD, Tashkin DP. Acute and chronic effects of cocaine on the immune system and the possible link to AIDS.J Neuroimmunol 1998;83(1–2):133–138.
131.Xu W, Flick T, Mitchel J, et al. Cocaine effects on immunocompetent cells: an observation of in vitro cocaine exposure. Int J Immunopharmacol 1999;21(7):463–472.
132.Baldwin GC, Buckley DM, Roth MD, et al. Acute activation of circulating polymorphonuclear neutrophils following in vivo administration of cocaine. A potential etiology for pulmonary injury. Chest1997;111(3):698–705.
133.Riezzo I, Fiore C, De Carlo D, et al. Side effects of cocaine abuse: multiorgan toxicity and pathological consequences. Curr Med Chem 2012;19(33):5624–5646.
134.Albertson TE, Walby WF, Derlet RW. Stimulant-induced pulmonary toxicity. Chest 1995;108(4):1140–1149.
135.Alnas M, Altayeh A, Zaman M. Clinical course and outcome of cocaine-induced pneumomediastinum. Am J Med Sci 2010;339(1):65–67.
136.Meleca RJ, Burgio DL, Carr RM, et al. Mucosal injuries of the upper aerodigestive tract after smoking crack or freebase cocaine. Laryngoscope 1997;107(5):620–625.
137.Reino AJ, Lawson W. Upper airway distress in crack-cocaine users.Otolaryngol Head Neck Surg 1993;109(5):937–940.
138.Libby DM, Klein L, Altorki NK. Aspiration of the nasal septum: a new complication of cocaine abuse. Ann Intern Med 1992;116(7):567–568.
139.Armstrong M, Jr, Shikani AH. Nasal septal necrosis mimicking Wegener’s granulomatosis in a cocaine abuser. Ear Nose Throat J 1996;75(9):623–626.
140.Restrepo CS, Carrillo JA, Martinez S, et al. Pulmonary complications from cocaine and cocaine-based substances: imaging manifestations. Radiographics 2007;27(4):941–956.
141.Ettinger NA, Albin RJ. A review of the respiratory effects of smoking cocaine. Am J Med 1989;87(6):664–668.
142.Tashkin DP, Kleerup EC, Koyal SN, et al. Acute effects of inhaled and i.v. cocaine on airway dynamics. Chest 1996;110(4):904–910.
143.Sirvent AE, Enriquez R, Andrada E, et al. Goodpasture’s syndrome in a patient using cocaine—a case report and review of the literature. Clin Nephrol 2007;68(3):182–185.
144.McGrath MM, Isakova T, Rennke HG, et al. Contaminated cocaine and antineutrophil cytoplasmic antibody-associated disease. Clin J Am Soc Nephrol 2011;6(12):2799–2805.
145.Smith WS, Matthay MA. Evidence for a hydrostatic mechanism in human neurogenic pulmonary edema. Chest 1997;111(5):1326–1333.
146.Hurst RT, Prasad A, Askew JW III, et al. Takotsubo cardiomyopathy: a unique cardiomyopathy with variable ventricular morphology.J Am Coll Cardiol Imaging 2010;3(6):641–649.
147.Kleerup EC, Wong M, Marques-Magallanes JA, et al. Acute effects of intravenous cocaine on pulmonary artery pressure and cardiac index in habitual crack smokers. Chest1997;111(1):30–35.
148.Smith GT, McClaughry PL, Purkey J, et al. Crack cocaine mimicking pulmonary embolism on pulmonary ventilation/perfusion lung scan. A case report. Clin Nucl Med 1995;20(1):65–68.
149.Delaney K, Hoffman RS. Pulmonary infarction associated with crack cocaine use in a previously healthy 23-year-old woman. Am J Med 1991;91(1):92–94.
150.Karch SB, Mari F, Bartolini V, et al. Aminorex poisoning in cocaine abusers. Int J Cardiol 2012;158(3):344–346.
151.Murray RJ, Smialek JE, Golle M, et al. Pulmonary artery medial hypertrophy in cocaine users without foreign particle microembolization. Chest 1989;96(5):1050–1053.
152.Nadeem S, Nasir N, Israel RH. Loffler’s syndrome secondary to crack cocaine. Chest 1994;105(5):1599–1600.
153.Kissner DG, Lawrence WD, Selis JE, et al. Crack lung: pulmonary disease caused by cocaine abuse. Am Rev Respir Dis 1987;136(5): 1250–1252.
154.Forrester JM, Steele AW, Waldron JA, et al. Crack lung: an acute pulmonary syndrome with a spectrum of clinical and histopathologic findings. Am Rev Respir Dis 1990;142(2): 462–467.
155.Orriols R, Munoz X, Ferrer J, et al. Cocaine-induced Churg-Strauss vasculitis. Eur Respir J 1996;9(1):175–177.
156.Drent M, Wijnen P, Bast A. Interstitial lung damage due to cocaine abuse: pathogenesis, pharmacogenomics and therapy. Curr Med Chem 2012;19(33):5607–5611.
157.Bailey ME, Fraire AE, Greenberg SD, et al. Pulmonary histopathology in cocaine abusers. Hum Pathol 1994;25(2): 203–207.
158.Albertson TE, Derlet RW, Van Hoozen BE. Methamphetamine and the expanding complications of amphetamines. West J Med 1999;170(4):214–219.
159.Touya JJ, Rahimian J, Corbus HF, et al. The lung as a metabolic organ. Semin Nucl Med 1986;16(4):296–305.
160.Pacifici R, Zuccaro P, Hernandez Lopez C, et al. Acute effects of 3,4-methylenedioxymethamphetamine alone and in combination with ethanol on the immune system in humans. J Pharmacol Exp Ther2001;296(1):207–215.
161.Lehnert BE, Ferin J. Particle binding, phagocytosis, and plastic substrate adherence characteristics of alveolar macrophages from rats acutely treated with chlorphentermine. J Reticuloendothel Soc1983;33(4):293–303.
162.Carvalho M, Carmo H, Costa VM, et al. Toxicity of amphetamines: an update. Arch Toxicol 2012;86(8):1167–1231.
163.Onwudike M. Ecstasy induced retropharyngeal emphysema. J Accid Emerg Med 1996;13(5):359–361.
164.Call TD, Hartneck J, Dickinson WA, et al. Acute cardiomyopathy secondary to intravenous amphetamine abuse. Ann Intern Med 1982;97(4):559–560.
165.Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med1996;335(9):609–616.
166.Chin KM, Channick RN, Rubin LJ. Is methamphetamine use associated with idiopathic pulmonary arterial hypertension? Chest 2006;130(6):1657–1663.
167.Fishman AP. Aminorex to fen/phen: an epidemic foretold. Circulation 1999;99(1):156–161.
168.Lewman LV. Fatal pulmonary hypertension from intravenous injection of methylphenidate (Ritalin) tablets. Hum Pathol 1972;3(1):67–70.
169.Schaiberger PH, Kennedy TC, Miller FC, et al. Pulmonary hypertension associated with long-term inhalation of “crank” methamphetamine. Chest 1993;104(2):614–616.
170.Herve P, Launay JM, Scrobohaci ML, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med 1995;99(3):249–254.
171.Schmidt RA, Glenny RW, Godwin JD, et al. Panlobular emphysema in young intravenous Ritalin abusers. Am Rev Respir Dis 1991;143(3):649–656.
172.Fligner CL, Opheim KE. Caffeine and its dimethylxanthine metabolites in two cases of caffeine overdose: a cause of falsely elevated theophylline concentrations in serum. J Anal Toxicol1988;12(6):339–343.
173.Seifert SM, Schaechter JL, Hershorin ER, et al. Health effects of energy drinks on children, adolescents, and young adults. Pediatrics 2011;127(3):511–528.
174.Zebraski SE, Kochenash SM, Raffa RB. Lung opioid receptors: pharmacology and possible target for nebulized morphine in dyspnea. Life Sci 2000;66(23):2221–2231.
175.Jaffe J. Goodman and Gilman’s the pharmacological basis of therapeutics. 8th ed. Oxford, UK: Pergamon Press, 1990.
176.Barke KE, Hough LB. Opiates, mast cells and histamine release. Life Sci 1993;53(18):1391–1399.
177.Eisenstein TK, Hilburger ME. Opioid modulation of immune responses: effects on phagocyte and lymphoid cell populations. J Neuroimmunol 1998;83(1–2):36–44.
178.Peterson PK, Molitor TW, Chao CC. The opioid-cytokine connection. J Neuroimmunol 1998;83(1–2):63–69.
179.Grimm MC, Ben-Baruch A, Taub DD, et al. Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med1998;188(2):317–325.
180.Yin D, Mufson RA, Wang R, et al. Fas-mediated cell death promoted by opioids. Nature 1999;397(6716):218.
181.Rosenow EC. Drug-induced pulmonary disease. Dis Mon 1994;40(5):258–310.
182.Krantz AJ, Hershow RC, Prachand N, et al. Heroin insufflation as a trigger for patients with life-threatening asthma. Chest 2003;123(2):510–517.
183.Yamanaka T, Sadikot RT. Opioid effect on lungs. Respirology 2013;18(2):255–262.
184.Moneo I, Alday E, Ramos C, Curiel G. Occupational asthma caused by Papaver somniferum. Allergol Immunopathol (Madr) 1993;21(4):145–148.
185.O’Donnell AE, Pappas LS. Pulmonary complications of intravenous drug abuse. Experience at an inner-city hospital. Chest 1988;94(2):251–253.
186.Sterrett C, Brownfield J, Korn CS, et al. Patterns of presentation in heroin overdose resulting in pulmonary edema. Am J Emerg Med 2003;21(1):32–34.
187.Cooper JA, Jr., White DA, Matthay RA. Drug-induced pulmonary disease. Part 2: Noncytotoxic drugs. Am Rev Respir Dis 1986;133(3):488–505.
188.Hakim TS, Grunstein MM, Michel RP. Opiate action in the pulmonary circulation. Pulm Pharmacol 1992;5(3):159–165.
189.Webster LR, Choi Y, Desai H, et al. Sleep-disordered breathing and chronic opioid therapy. Pain Med 2008;9(4):425–432.
190.Wang D, Teichtahl H, Drummer O, et al. Central sleep apnea in stable methadone maintenance treatment patients. Chest 2005;128(3):1348–1356.
191.Teichtahl H, Wang D, Cunnington D, et al. Ventilatory responses to hypoxia and hypercapnia in stable methadone maintenance treatment patients. Chest 2005;128(3):1339–1347.
192.Sharkey KM, Kurth ME, Anderson BJ, et al. Obstructive sleep apnea is more common than central sleep apnea in methadone maintenance patients with subjective sleep complaints. Drug Alcohol Depend2010;108(1–2):77–83.
193.Karne S, D’Ambrosio C, Einarsson O, et al. Hypersensitivity pneumonitis induced by intranasal heroin use. Am J Med 1999;107(4):392–395.
194.Heinemann HO. Alcohol and the lung. A brief review. Am J Med 1977;63(1):81–85.
195.Scanlan MF, Roebuck T, Little PJ, et al. Effect of moderate alcohol upon obstructive sleep apnoea. Eur Respir J 2000;16(5):909–913.
196.Cook RT. Alcohol abuse, alcoholism, and damage to the immune system—a review. Alcohol Clin Exp Res 1998;22(9):1927–1942.
197.Adams HG, Jordan C. Infections in the alcoholic. Med Clin North Am 1984;68(1):179–200.
198.Boe DM, Vandivier RW, Burnham EL, et al. Alcohol abuse and pulmonary disease. J Leukoc Biol 2009;86(5):1097–1104.
199.Mehta AJ, Guidot DM. Alcohol abuse, the alveolar macrophage and pneumonia. Am J Med Sci 2012;343(3):244–247.
200.Wrenn KD, Slovis CM, Minion GE, et al. The syndrome of alcoholic ketoacidosis. Am J Med 1991;91(2):119–128.
201.Krasowski MD, Wilcoxon RM, Miron J. A retrospective analysis of glycol and toxic alcohol ingestion: utility of anion and osmolal gaps. BMC Clin Pathol 2012;12(1):1.
202.Kraut JA, Kurtz I. Toxic alcohol ingestions: clinical features, diagnosis, and management. Clin J Am Soc Nephrol 2008;3(1):208–225.
203.Lustik SJ, Chhibber AK, Kolano JW, et al. The hyperventilation of cirrhosis: progesterone and estradiol effects. Hepatology 1997;25(1):55–58.
204.Linneberg A, Berg ND, Gonzalez-Quintela A, et al. Prevalence of self-reported hypersensitivity symptoms following intake of alcoholic drinks. Clin Exp Allergy 2008;38(1):145–151.
205.Lieberoth S, Backer V, Kyvik KO, et al. Intake of alcohol and risk of adult-onset asthma. Respir Med 2012;106(2):184–188.
206.Vally H, Carr A, El-Saleh J, et al. Wine-induced asthma: a placebo-controlled assessment of its pathogenesis. J Allergy Clin Immunol 1999;103(1 Pt 1):41–46.
207.Siu ST, Udaltsova N, Iribarren C, et al. Alcohol and lung airways function. Perm J 2010;14(1):11–18.
208.Fujimura M, Myou S, Kamio Y, et al. Increased airway responsiveness to acetaldehyde in asthmatic subjects with alcohol-induced bronchoconstriction. Eur Respir J 1999;14(1):19–22.
209.Gong H, Jr., Tashkin DP, Calvarese BM. Alcohol-induced bronchospasm in an asthmatic patient: pharmacologic evaluation of the mechanism. Chest 1981;80(2):167–173.
210.Shimoda T, Kohno S, Takao A, et al. Investigation of the mechanism of alcohol-induced bronchial asthma. J Allergy Clin Immunol 1996;97(1 Pt 1):74–84.
211.Krok KL, Cárdenas A. Hepatic hydrothorax. Semin Resp Crit Care Med 2012;33(1):03–10.
212.Jacinto J. Hepatic Hydrothorax, Clinical Characteristics, and Surgical Treatment Results. Chest. 2012;142(4_MeetingAbstracts):66A.
213.Kilburn JP, Hutchings J, Misselhorn D, et al. Use of indwelling tunneled pleural catheters for the management of hepatic hydrothorax. Chest 2010;138(4 MeetingAbstracts):418A-A.
214.Rossle M, Gerbes AL. TIPS for the treatment of refractory ascites, hepatorenal syndrome and hepatic hydrothorax: a critical update. Gut 2010;59(7):988–1000.
215.Yao EH, Kong BC, Hsue GL, et al. Pulmonary function changes in cirrhosis of the liver. Am J Gastroenterol 1987;82(4):352–354.
216.King PD, Rumbaut R, Sanchez C. Pulmonary manifestations of chronic liver disease. Dig Dis 1996;14(2):73–82.
217.Kochar R, Nevah Rubin MI, Fallon MB. Pulmonary complications of cirrhosis. Curr Gastroenterol Rep 2011;13(1):34–39.
218.Kuo PC, Plotkin JS, Gaine S, et al. Portopulmonary hypertension and the liver transplant candidate. Transplantation 1999;67(8):1087–1093.
219.Van der Linden P, Le Moine O, et al. Pulmonary hypertension after transjugular intrahepatic portosystemic shunt: effects on right ventricular function. Hepatology 1996;23(5):982–987.
220.Moss M, Guidot DM, Wong-Lambertina M, et al. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med 2000;161(2 Pt 1):414–419.
221.Wood DM, Brailsford AD, Dargan PI. Acute toxicity and withdrawal syndromes related to gamma-hydroxybutyrate (GHB) and its analogues gamma-butyrolactone (GBL) and 1,4-butanediol (1,4-BD). Drug Test Anal 2011;3(7–8):417–425.
222.Sporer KA, Chin RL, Dyer JE, et al. Gamma-hydroxybutyrate serum levels and clinical syndrome after severe overdose. Ann Emerg Med 2003;42(1):3–8.
223.Bronstein AC, Spyker DA, Cantilena LR, Jr, et al. 2007 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 25th Annual Report. Clin Toxicol (Phila)2008;46(10):927–1057.
224.Calcaterra S, Glanz J, Binswanger IA. National trends in pharmaceutical opioid related overdose deaths compared to other substance related overdose deaths: 1999–2009. Drug Alcohol Depend2013;131(3):263–270.
225.Graudins A AC. Intensive care medicine. Boston, MA: Little, Brown, Company, 1999.
226.Alkhouri I, Gibbons P, Ravindranath D, et al. Substance-related psychiatric emergencies. In: Riba MB, Ravindranath D: Clinical Manual of Emergency Psychiatry. Washington, DC: American Psychiatric Publishing, 2010:187–206.
227.Kuzniar TJ, Balagani R, Radigan KA, et al. Coma with absent brainstem reflexes resulting from zolpidem overdose. Am J Ther 2010;17(5):e172–e174.
228.Roberts DM, Buckley NA. Enhanced elimination in acute barbiturate poisoning - a systematic review. Clin Toxicol (Phila) 2011;49(1):2–12.
229.Marin M, Riaza MD, Quintana A, et al. P03-219—New agents for the benzodiazepine withdrawal syndrome. Eur Psychiatry 2010;25(0):1286.
230.Kuiper MA, Peikert N, Boerma EC. Gamma-hydroxybutyrate withdrawal syndrome: a case report. Cases J 2009;2:6530.
231.Jangir SN, Suthar R, Deshpande SN. Two cases of seizures on sudden withdrawal of supra-therapeutic doses of zolpidem (selective omega I benzodiazepine receptor agonist). Int J Case Rep Images (IJCRI) 2012;3(4):1–4.
232.Guilleminault C. Benzodiazepines, breathing, and sleep. Am J Med 1990;88(3A):25S–28S.
233.Cirignotta F, Mondini S, Zucconi M, et al. Zolpidem-polysomnographic study of the effect of a new hypnotic drug in sleep apnea syndrome. Pharmacol Biochem Behav 1988;29(4):807–809.
234.George CF, Feldman N, Inhaber N, et al. A safety trial of sodium oxybate in patients with obstructive sleep apnea: acute effects on sleep-disordered breathing. Sleep Med2010;11(1):38–42.
235.Marsolek MR, White NC, Litovitz TL. Inhalant abuse: monitoring trends by using poison control data, 1993–2008. Pediatrics 2010;125(5):906–913.
236.MacIver MB. Abused inhalants enhance GABA-mediated synaptic inhibition. Neuropsychopharmacology 2009;34(10):2296–2304.
237.Flanagan RJ, Ives RJ. Volatile substance abuse. Bull Narc 1994;46(2):49–78.
238.Linden CH. Volatile substances of abuse. Emerg Med Clin North Am 1990;8(3):559–578.
239.Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999;34(5):646–656.
240.Fischman CM. Toxic effects of toluene. JAMA 1979;242(14):1491.\
241.Redlich CA, Karol MH. Diisocyanate asthma: clinical aspects and immunopathogenesis. Int Immunopharmacol 2002;2(2–3): 213–224.
242.Budnik LT, Preisser AM, Permentier H, et al. Is specific IgE antibody analysis feasible for the diagnosis of methylenediphenyl diisocyanate-induced occupational asthma? Int Arch Occup Environ Health2013;86:417–430.
243.Baur X. Hypersensitivity pneumonitis (extrinsic allergic alveolitis) induced by isocyanates. J Allergy Clin Immunol 1995;95(5 Pt 1):1004–1010.
244.Covalla JR, Strimlan CV, Lech JG. Severe tracheobronchitis from inhalation of an isobutyl nitrite preparation. Drug Intell Clin Pharm 1981;15(1):51–52.
245.Brouette T, Anton R. Clinical review of inhalants. Am J Addict 2001;10(1):79–94.
246.LiPuma JP, Wellman J, Stern HP. Nitrous oxide abuse: a new cause of pneumomediastinum. Radiology 1982;145(3):602.
247.Wagner SA, Clark MA, Wesche DL, et al. Asphyxial deaths from the recreational use of nitrous oxide. J Forensic Sci 1992;37(4):1008–1015.
248.Montine TJ. Massive pulmonary embolus and anabolic steroid abuse. JAMA 1992;267(17):2328.
249.Akhter J, Hyder S, Ahmed M. Cerebrovascular accident associated with anabolic steroid use in a young man. Neurology 1994;44(12):2405–2406.
250.Ferenchick GS, Hirokawa S, Mammen EF, et al. Anabolic-androgenic steroid abuse in weight lifters: evidence for activation of the hemostatic system. Am J Hematol 1995;49(4):282–288.
251.Rochester CL, Mohsenin V. Respiratory complications of stroke. Semin Resp Crit Care Med 2002;23:248–260.