Alex Sparreboom and Sharyn D. Baker
INTRODUCTION
Drug selection and therapy considerations in oncology were originally solely based on observations of the effects produced.1 To overcome some of the limitations of this empirical approach and to answer questions related to considerations of dose, frequency, and duration of drug treatment, it is necessary to understand the events that follow drug administration. Preclinical in vitro and in vivo studies have shown that the magnitude of antitumor response is a function of the concentration of drug,2 and this has led to the suggestion that the therapeutic objective can be achieved by maintaining an adequate concentration at the site of action for the duration of therapy.3 However, drugs are rarely directly administered at their sites of action. Indeed, most anticancer drugs are given intravenously or orally, and yet are expected to act in the brain, lungs, or elsewhere. Drugs must, therefore, move from the site of administration to the site of action and, moreover, distribute to all other tissues including organs that eliminate them from the body, such as the kidneys and liver. To administer drugs optimally, knowledge is needed not only of the mechanisms of drug absorption, distribution, and elimination, but also of the kinetics of these processes.4
The treatment of human malignancies involving drugs can be divided into two pharmacologic phases, a pharmacokinetic phase in which the dose, dosage form, frequency, and route of administration are related to drug level–time relationships in the body, and a pharmacodynamic phase in which the concentration of drug at the site(s) of action is related to the magnitude of the effect(s) produced. Once both of these phases have been defined, a dosage regimen can be designed to achieve the therapeutic objective, although additional factors need to be taken into consideration (Fig. 15.1). The clinical application of this approach allows distinctions between pharmacokinetic and pharmacodynamic causes of an unusual drug response. A basic tenet of pharmacokinetics is that the magnitude of both the desired response and toxicity are functions of the drug concentration at the site(s) of action. Accordingly, therapeutic failure results when either the concentration is too low, resulting in ineffective therapy, or is too high, producing unacceptable toxicity. Between these limits of concentrations lies a region associated with therapeutic success, the so-called therapeutic window.5 Because the concentration of a drug at the site of action can rarely be measured directly, with the exception of certain hematologic malignancies, plasma or blood is commonly measured instead as a more accessible alternative.

PHARMACOKINETIC CONCEPTS
A drug’s pharmacokinetic properties can be defined by two fundamental processes affecting drug behavior over time, absorption and disposition.
Absorption
Historically, most anticancer drugs have been administered intravenously; however, the use of orally administered agents is growing with the development of small-molecule targeted cancer therapeutics, such as tyrosine kinase inhibitors.6 Moreover, drugs may also be administered regionally, for example into the pleural or peritoneal cavities,7 the cerebrospinal fluid, or intra-arterially into a vessel leading to a cancerous tissue.8 The process by which the unchanged drug moves from the site of administration to the site of measurement within the body is referred to as absorption. Loss at any site prior to the site of measurement contributes to a decrease in the apparent absorption of a drug. For an orally administered agent, this complex series of events involves disintegration of the pharmaceutical dosage form, dissolution, diffusion through gastrointestinal fluids, permeation of the gut membrane, portal circulation uptake, passage through the liver, and, finally, entry into the systemic circulation. The loss of drug as it passes for the first time through organs of elimination, such as the gastrointestinal membranes and the liver, during the absorption process is known as the first-pass effect.9
The pharmacokinetic parameter most closely associated with absorption is availability or bioavailability (F), defined as the fraction (or percent) of the administered dose that is absorbed intact. Bioavailability can be estimated by dividing the area under the plasma concentration–time curve (AUC) achieved following extravascular administration by the AUC observed after intravenous administration, and can range from 0 to 1.0 (or 0% to 100%).
Disposition
Disposition is defined as all the processes that occur subsequent to absorption of a drug; by definition, the components of disposition are distribution and elimination. Distribution is the process of reversible transfer of a drug to and from the site of measurement. Any drug that leaves the site of measurement and does not return has undergone elimination, which occurs by two processes, excretion and metabolism. Excretion is the irreversible loss of the chemically unchanged drug, whereas metabolism is the conversion of drug to another chemical species.
The extent of drug distribution can be determined by relating the concentration obtained with a known amount of drug in the body and is, in essence, a dilution space. The apparent volume into which a drug distributes in the body at equilibrium in called the volume of distribution (Vd), and may or may not correspond to an actual physiologic compartment.
The rate and extent to which a drug distributes into various tissues depend on a number of factors, including hydrophobicity, tissue permeability, tissue-binding constants, binding to serum proteins, and local organ blood flow.10Large apparent volumes of distribution are common for agents with high tissue binding or high lipid solubility, although distribution into specific body compartments may be limited by physiologic processes, such as the blood–brain barrier protecting the central nervous system11,12 or the blood–testes barrier.13
Just as Vd is needed as a parameter to relate the concentration to the amount of drug in the body, there is also a need to have a parameter to relate the concentration to the rate of drug elimination, which is known as clearance (CL). Of all pharmacokinetic parameters, CL has the most clinical relevance because it defines the key relationship between drug dose and systemic drug exposure (AUC). Derived from Vd and CL is the parameter elimination rate constant, which can be regarded as the fractional rate of drug removal. It is, however, more common to refer to the half-life than to the elimination rate constant of a drug. The half-life of a drug is a useful parameter to estimate the time required to reach steady state on a multidose schedule or during a continuous intravenous drug infusion.
Dose Proportionality
When drug concentrations change in strict proportionality to the dose of drug administered, then the condition of dose proportionality (or linear pharmacokinetics) holds. If doubling the dose exactly doubles the plasma concentration or AUC, then pharmacokinetic parameters such Vd, and CL are constant and remain independent of dose and concentration.14 By strict definition, drugs with linear pharmacokinetics are dose proportional. Dose proportionality is clinically important because it means that dose adjustments will generate predictable changes in systemic drug exposure. For drugs that lack dose proportionality, Vd and CL will demonstrate concentration or time dependence, or both, making it difficult to predict the effect of dose adjustments on drug concentration (Fig. 15.2). Factors that can contribute to a lack of dose proportional pharmacokinetics include saturable oral absorption,15 capacity-limited distribution or protein binding,16 and/or saturable metabolism.17 Dose proportionality of anticancer agents is typically assessed in Phase 1 dose-escalation trials in which small groups of patients are treated at a single dose level using a parallel study design, although the statistical power of such studies to detect deviations from dose proportionality is poor. An alternative, more robust study design is a crossover study in which each patient receives a low dose, an intermediate dose, and a high dose over consecutive cycles of treatment.18 However, such studies are relatively rare in oncology because of the required use of low, potentially ineffective doses, which may raise ethical concerns for patients.

PHARMACODYNAMIC CONCEPTS
Pharmacodynamic models relate clinical drug effects with drug dose, concentration, or other pharmacokinetic parameters indicative of drug exposures (Table 15.1). In oncology, pharmacodynamic variability may account for substantial differences in clinical outcomes, even when systemic exposures are uniform. Variability in pharmacodynamic response may be heavily influenced by clinical covariates such as age, gender, prior chemotherapy, prior radiotherapy, concomitant medications, or other variables.19 The pharmacokinetic parameters that are most often correlated with drug effects are markers of drug exposure, such as AUC. In general, the specific parameter used as the independent variable in a pharmacodynamic analysis depends on the particular characteristics of the study drug.

In oncology, pharmacodynamic studies of drug effects have most often focused on toxicity endpoints.20 Continuous response variables, such as the percentage fall in the absolute blood count from baseline, are easily analyzed using nonlinear regression methods. Dose-limiting neutropenia has been frequently analyzed using a sigmoid maximum effect model described by the modified Hill equation. The pharmacodynamic analysis of subjectively graded clinical endpoints, such as common toxicity criteria scores on a 4-point scale, may require more sophisticated statistical methods.21,22 Logistical regression methods have been used to model these types of categorical (ordinal) response or outcome variables.
Physiologic pharmacodynamic models describing the severity and time course of drug-related myelosuppression have been derived using population mixed-effect methods for several agents, including paclitaxel23,24 and pemetrexed.25 The ability of these models to predict both the severity and duration of drug-induced neutropenia substantially enhances their clinical usefulness.26 In contrast to small-molecule therapeutics, large-molecule therapeutics such as monoclonal antibodies may not demonstrate toxicities directly related to dose levels. For these agents, a thorough understanding of the pharmacokinetic/pharmacodynamic relationships using modeling approaches may be critical for optimal dose selection.27
The antitumor activity of certain chemotherapeutic agents is highly schedule dependent. For such drugs, the dose fractionated over several days can produce a different antitumor response or toxicity profile compared with the same dose given over a shorter period. For example, the efficacy of etoposide in the treatment of small-cell lung cancer is markedly increased when an identical total dose of etoposide is administered by a 5-day divided-dose schedule rather than a 24-hour infusion.28 Pharmacokinetic analysis in that study showed that both schedules produced very similar overall drug exposure (as measured by AUC), but that the divided-dose schedule produced twice the duration of exposure to an etoposide plasma concentration of >1 μg/mL. This finding has led to the use of prolonged oral administration of etoposide to treat patients with cancer.29 Similar schedule dependence has been demonstrated for a number of other anticancer agents, notably paclitaxel30,31 and topotecan.32 For these agents, the variability in clinically tested treatment schedules is enormous, ranging from short intravenous infusions of less than 30 minutes to 21-day or even 7-week continuous infusion administrations, with large differences in experienced toxicity profiles.
VARIABILITY IN PHARMACOKINETICS/PHARMACODYNAMICS
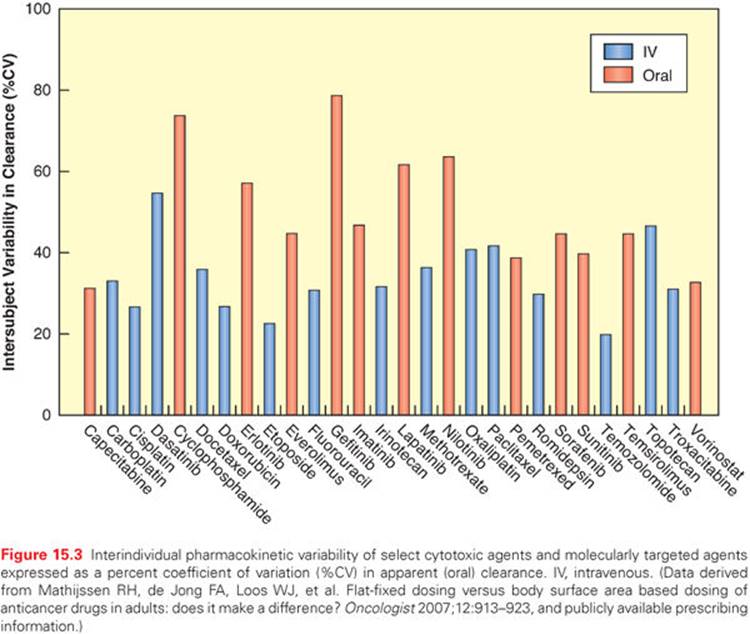
There is often a marked variation in drug handling between individual patients, resulting in variability in pharmacokinetic parameters (Fig. 15.3), which will often lead to variability in the pharmacodynamic effects of a given dose of a drug.33 That is, an identical dose of drug may result in acceptable toxicity in one patient, and unacceptable and possibly life-threatening toxicity in another, or a clinical response in one individual and cancer progression in another. The principal underlying sources of this interindividual pharmacokinetic/pharmacodynamic variability are discussed in the following paragraphs.
Body Size and Body Composition
The traditional method of individualizing anticancer drug dosage is by using body surface area (BSA).34 However, the usefulness of normalizing an anticancer drug dose to BSA in adults has been questioned, because, for many drugs, there is no relationship between BSA and CL.35 Likewise, attempts to replace BSA as a size metric in dose calculation with alternate descriptors such as lean body weight, either in an average population or in individuals at the outer extremes of weight (i.e., frail, severely obese patients) have failed for many anticancer agents.36,37 It should be pointed out that BSA is a much more important consideration in drug dose calculation for pediatric patients as compared to adults, because of the larger size range in the former population.38 Based in part on the failure to reduce interindividual pharmacokinetic variability with the use of BSA normalization to obtain a starting dose, many of the more recently developed molecularly targeted agents are currently administered using a flat-fixed dose irrespective of an individual’s BSA.37
Age
Changes in body composition and organ function at the extremes of age can affect both drug disposition and drug effect.39 For example, maturational processes in infancy may alter the absorption and distribution of drugs as well as change the capacity for drug metabolism and excretion.4 The importance of understanding the influence of age on the pharmacokinetics and pharmacodynamics of individual anticancer agents has increased steadily as treatment for the malignancies of infants,40 adolescents,41 and the elderly42 has advanced. Although pediatric cancers remain rare compared with cancers in adults and the elderly population, in particular, optimizing treatment in a patient group with a high cure rate and a long expected survival becomes critical to minimize the incidence of preventable late complications while maintaining efficacy.
Pathophysiologic Changes
Effects of Disease
Pathophysiologic changes associated with particular malignancies may cause dramatic alterations in drug disposition. For example, increases in the clearance of both antipyrine and lorazepam were noted after remission induction compared with the time of diagnosis in children with acute lymphoblastic leukemia (ALL).43 The clearance of unbound teniposide is lower in children with ALL in relapse than during first remission.44 Because leukemic infiltration of the liver at the time of diagnosis is common, drugs metabolized by the liver may have a reduced clearance, as has been documented in preclinical models.45
Furthermore, in mouse models, certain tumors elicited an acute phase response that coincided with downregulation of human CYP3A4 in the liver as well as the mouse ortholog Cyp3a11.46 The reduction of murine hepatic Cyp3a gene expression in tumor-bearing mice resulted in decreased Cyp3a protein expression and, consequently, a significant reduction in Cyp3a-mediated metabolism of midazolam. These findings support the possibility that tumor-derived inflammation may alter the pharmacokinetic and pharmacodynamic properties of CYP3A4 substrates, leading to reduced metabolism of drugs in humans.47This supports a possible need for disease-specific design of early clinical trials with anticancer drugs,48 as has been recommended for docetaxel.49
Effects of Renal Impairment
The potential impact of pathophysiologic status on interindividual pharmacokinetic variability can be due to either the disease itself or to a dysfunction of specific organs involved in drug elimination. For example, if urinary excretion is an important elimination route for a given drug, any decrement in renal function could lead to decreased drug clearance, which may result in drug accumulation and toxicity.50Therefore, it would be logical to decrease the drug dose relative to the degree of impaired renal function in order to maintain plasma concentrations within a target therapeutic window. The best known example of this a priori dose adjustment of an anticancer agent remains carboplatin, which is excreted renally almost entirely by glomerular filtration. Various strategies have been developed to estimate carboplatin doses based on renal function among patients, either using creatinine clearance51 or glomerular filtration rates as measured by a radioisotope method.52 The application of these procedures has led to a substantial reduction in pharmacokinetic variability, such that carboplatin is currently one of the few drugs routinely administered to achieve a target exposure rather than on a milligram per square meter or milligram per kilogram basis.
The U.S. Food and Drug Administration (FDA) has developed a guidance on the impact of renal impairment on the pharmacokinetics, dosing, and labeling of drugs.53 The impact of this guidance has been assessed following a survey of 94 new drug applications for small-molecule new molecular entities approved over the years 2003 to 2007. The survey results indicated that 41% of the applications that included renal impairment study data resulted in a recommendation of dose adjustment in renal impairment.54 Interestingly, the survey results provided evidence that renal impairment can affect the pharmacokinetics of drugs that are predominantly eliminated by nonrenal processes such as metabolism and/or active transport. The latter finding supports the FDA recommendation to evaluate pharmacokinetic/pharmacodynamic alterations in renal impairment for those drugs that are predominantly eliminated by nonrenal processes, in addition to those that are mainly excreted unchanged by the kidneys. A striking example of a drug in the former category is imatinib, an agent that is predominantly eliminated by hepatic pathways but where predialysis renal impairment is associated with dramatically reduced drug clearance,55 presumably due to a transporter-mediated process.56
Effects of Hepatic Impairment
In contrast to the predictable decline in renal clearance of drugs when glomerular filtration is impaired, it is difficult to make general predictions on the effect of impaired liver function on drug clearance. The major problem is that commonly applied criteria to establish hepatic impairment are typically not good indicators of drug-metabolizing enzyme activity and that several alternative hepatic function tests, such as indocyanine green and antipyrine, have relatively limited value in predicting anticancer drug pharmacokinetics. An alternative dynamic measure of liver function has been proposed, which is based on totaled values (scored to the World Health Organization [WHO] grading system) of serum bilirubin, alkaline phosphatase, and either alanine aminotransferase or aspartate aminotransferase to give a hepatic dysfunction score.57 Based on pharmacokinetic studies in patients with normal and impaired hepatic function, guidelines have been proposed for dose adjustments of several agents when administered to patients with severe liver dysfunction.58 It should be emphasized that no uniform criteria have been used in the conduct of these studies and that, ultimately, substantial advances could be made through an a priori determination of the hepatic activity of enzymes of pertinent relevance to the chemotherapeutic drug(s) of interest, as has been done for docetaxel.59
Effects of Serum Proteins
The binding of drugs to serum proteins, particularly those that are highly bound, may also have significant clinical implications for a therapeutic outcome.60 Although protein binding is a major determinant of drug action, it is clearly only one of a myriad of factors that influence the disposition of anticancer drugs.16 The extent of protein binding is a function of drug and protein concentrations, the affinity constants for the drug–protein interaction, and the number of protein-binding sites per class of binding site. Because only the unbound (or free) drug in plasma water is available for distribution, the therapeutic response will correlate with free drug concentration rather than total drug concentration. Several clinical situations, including liver and renal disease, can significantly decrease the extent of serum binding and may lead to higher free drug concentrations and a possible risk of unexpected toxicity, although the total (free plus bound forms) plasma drug concentrations are unaltered.61 It is important to realize, however, that after therapeutic doses of most anticancer drugs, binding to serum proteins is independent of drug concentration, suggesting that the total plasma concentration is reflective of the unbound concentration. For some anticancer agents, including etoposide62 and paclitaxel,63 however, protein binding is highly dependent on dose and schedule.
Sex Dependence
A number of pharmacokinetic analyses have suggested that male gender is positively correlated with the maximum elimination capacity of various anticancer drugs (e.g., paclitaxel)8 or with increased clearance (e.g., imatinib)64compared with female gender. These observations have added to a growing body of evidence that the pharmacokinetic profile of various anticancer drugs exhibits significant sexual dimorphism, which is rarely considered in the design of clinical trials during oncology drug development.
Drug Interactions
Coadministration of Other Chemotherapeutic Drugs
Favorable and unfavorable interactions between drugs must be considered in developing combination regimens. These interactions may influence the effectiveness of each of the components of the combination, and typically occur when the pharmacokinetic profile of one drug is altered by the other. Such interactions are important in the design of trials evaluating drug combinations because, occasionally, the outcome of concurrent drug administration is diminished therapeutic efficacy or increased toxicity of one or more of the administered agents. Although a recent survey indicated that clinically significant pharmacokinetic interactions are relatively rare in Phase I trials of oncology drug combinations,65 interactions appear to be more common for combinations of tyrosine kinase inhibitors with cytotoxic chemotherapeutics.66
Coadministration of Nonchemotherapeutic Drugs
Many prescription and over-the-counter medications have the potential to cause interactions with anticancer agents by altering their pharmacokinetic characteristics and leading to clinically significant phenotypes. Most clinically relevant drug interactions in this category are due to changes in metabolic routes related to an altered expression or function of cytochrome P450 (CYP) isozymes. This class of enzymes, particularly the CYP3A4 isoform, is responsible for the oxidation of a large proportion of currently approved anticancer drugs. Elevated CYP activity (induction), translated into a more rapid metabolic rate, may result in a decrease in plasma concentrations and to a loss of therapeutic effect. For example, anticonvulsant drugs such as phenytoin, phenobarbital, and carbamazepine can induce drug-metabolizing enzymes and thereby increase the clearance of various anticancer agents.33
Conversely, the suppression (inhibition) of CYP activity, for example with ketoconazole,13,67 may trigger a rise in plasma concentrations and can lead to exaggerated toxicity commensurate with overdose. It should be borne in mind that several pharmacokinetic parameters could be altered simultaneously. Especially in the development of anticancer agents given by the oral route, oral bioavailability plays a crucial role9; this parameter is contingent on adequate absorption and the circumvention of intestinal and, subsequently, hepatic metabolism of the drug. It has been suggested that the prevalence of drug–drug interactions is particularly high in cancer patients receiving oral chemotherapy,68 especially for agents that are weak bases that exhibit pH-dependent solubility.69
An additional consideration is related to a possible influence of food intake on the extent of drug absorption after oral administration, which can increase, decrease, or remain unchanged depending on specific physicochemical properties of the drug in question (Table 15.2). The relatively narrow therapeutic index of most of these agents means that significant inter- and intrapatient variability would predispose some individuals to excessive toxicity or, conversely, inadequate efficacy.12

Coadministration of Complementary and Alternative Medicine
Surveys within the past decade estimate the prevalence of complementary and alternative medicine (CAM) use in oncology patients to be as high as 87%, and in many cases the treating physician is not aware of the patients’ CAM use.70 With a larger number of participants to phase I clinical trials71 using herbal treatments combined with allopathic therapies, the risk for herb–drug interactions is a growing concern, and there is an increasing need to understand possible adverse drug interactions in oncology at the early stages of drug development.
A number of clinically important pharmacokinetic interactions involving CAM and cancer drugs have now been recognized, although causal relationships have not always been established.72 Most of the observed interactions point to the herbs affecting several isoforms of the CYP family, either through inhibition or induction. In the context of chemotherapeutic drugs, St. John’s wort,73 garlic,74 milk thistle,75and Echinacea11 have been formally evaluated for their pharmacokinetic drug–interaction potential in cancer patients. However, various other herbs have the potential to significantly modulate the expression and/or activity of drug-metabolizing enzymes and drug transporters (Table 15.3), including ginkgo, ginseng, and kava.70 Because of the high prevalence of herbal medicine use, physicians should include herb usage in their routine drug histories in order to have an opportunity to outline to individual patients which potential hazards should be taken into consideration prior to participation in a clinical trial.

Inherited Genetic Factors
The discipline of pharmacogenetics describes differences in the pharmacokinetics and pharmacodynamics of drugs as a result of inherited variation in drug metabolizing enzymes, drug transporters, and drug targets between patients.76 These inherited variations are occasionally responsible for extensive interpatient variability in drug exposure or effects. Severe toxicity might occur in the absence of a typical metabolism of active compounds, while the therapeutic effect of a drug could be diminished in the case of an absence of activation of a prodrug, such as irinotecan.77 The importance and detectability of polymorphisms for a given enzyme or transporter depends on the contribution of the variant gene product to pharmacologic response, the availability of alternative pathways of elimination, and the frequency of occurrence of the variant allele. Although many substrates have been identified for the known polymorphic drug metabolizing enzymes and transporters, the contribution of a genetically determined source of interindividual pharmacokinetic variability has been established for only a few cancer chemotherapeutic agents. Most of these cases involve agents for which elimination is critically dependent on a rate-limiting breakdown by a polymorphic enzyme (e.g., 6-mercaptopurine by thiopurine-S-methyltransferase; 5-fluorouracil by dihydropyrimidine dehydrogenase) or when a polymorphic enzyme is involved in the formation of a toxic metabolite (e.g., tamoxifen by CYP2D6).78
In addition to drug metabolism, pharmacokinetic processes are highly dependent on the interplay with drug transport in organs such as the intestines, kidneys, and liver. Genetically determined variation in drug transporter function or expression is now increasingly recognized to have a significant role as a determinant of intersubject variability in response to various commonly prescribed drugs.79 The most extensively studied class of drug transporters are those encoded by the family of ATP-binding cassette (ABC) genes, some of which also play a role in the resistance of malignant cells to anticancer agents. Among the 48 known ABC gene products, ABCB1 (P-glycoprotein), ABCC1 (multidrug-resistance associated protein-1 [MRP1]) and its homologue ABCC2 (MRP2; cMOAT), and ABCG2 (breast cancer resistance protein [BCRP]) are known to influence the oral absorption and disposition of a wide variety of drugs.80 As a result, the expression levels of these proteins in humans have important consequences for an individual’s susceptibility to certain anticancer drug–induced side effects, interactions, and treatment efficacy, for example, in the case of genetic variation in ABCG2 in relation to gefitinib-induced diarrhea.81
Similar to the discoveries of functional genetic variations in drug efflux transporters of the ABC family, there have been considerable advances in the identification of inherited variants in transporters that facilitate cellular drug uptake in tissues that play an important role in drug elimination, such as the liver (Fig. 15.4). Among these, members of the organic anion-transporting polypeptides (OATP), organic anion transporters (OAT), and organic cation transporters (OCT) can mediate the cellular uptake of a large number of structurally divergent compounds.82,83 Accordingly, functionally relevant polymorphisms in these influx transporters may contribute to interindividual and interethnic variability in drug disposition and response,84 for example, in the case of the impact of polymorphic variants in the OCT1 gene SLC22A1 on the survival of patients with chronic myeloid leukemia receiving treatment with imatinib.85

DOSE-ADAPTATION USING PHARMACOKINETIC/PHARMACODYNAMIC PRINCIPLES
Therapeutic Drug Monitoring
Prolonged infusion schedules of anticancer drugs offer a very convenient setting for dose adaptation in individual patients. At the time required to achieve steady-state concentration, it is possible to modify the infusion rate for the remainder of the treatment course if a relationship is known between this steady-state concentration and a desired pharmacodynamic endpoint. This method has been successfully used to adapt the dose during continuous infusions of 5-fluorouracil and etoposide, and for repeated oral administration of etoposide or repeated intravenous administration of cisplatin.86 Methotrexate plasma concentrations are routinely monitored to identify patients at high risk of toxicity and to adjust leucovorin rescue in patients with delayed drug excretion. This monitoring has significantly reduced the incidence of serious toxicity, including toxic death, and in fact, has improved outcome by eliminating unacceptably low systemic exposure levels.87 Therapeutic drug monitoring has also been applied to or is currently under investigation for several more recently developed anticancer drugs, including imatinib88–90 and sorafenib.91
Feedback-Controlled Dosing
It remains to be determined how information on interindividual pharmacokinetic variability can eventually be used to devise an optimal dosage regimen of a drug for the treatment of a given disease in an individual patient. Obviously, the desired objective would be most efficiently achieved if the individual’s dosage requirements could be calculated prior to administering the drug. While this ideal cannot be met completely in clinical practice, with the notable exception of carboplatin, some success may be achieved by adopting feedback-controlled dosing. In the adaptive dosage with feedback control, population-based predictive models are used initially, but allow the possibility of dosage alteration based on feedback revision. In this approach, patients are first treated with standard dose and, during treatment, pharmacokinetic information is estimated by a limited-sampling strategy and compared with that predicted from the population model with which treatment was initiated. On the basis of the comparison, more patient-specific pharmacokinetic parameters are calculated, and dosage is adjusted accordingly to maintain the target exposure measure producing the desired pharmacodynamic effect. Despite its mathematical complexity, this approach may be the only way to deliver the desired and precise exposure of an anticancer agent.
The study of population pharmacokinetics seeks to identify the measurable factors that cause changes in the dose-concentration relationship and the extent of these alterations so that, if these are associated with clinically significant shifts in the therapeutic index, dosage can be appropriately modified in the individual patient. It is obvious that a careful collection of data during the development of drugs and subsequent analyses could be helpful to collect some essential information on the drug. Unfortunately, important information is often lost by failing to analyze this data or due to the fact that the relevant samples or data were never collected. Historically, this has resulted in the notion that tools for the identification of patient population subgroups are inadequate for most of the currently approved anticancer drugs.
However, the use of population pharmacokinetic models is increasingly studied in an attempt to accommodate as much of the pharmacokinetic variability as possible in terms of measurable characteristics. This type of analysis has been conducted for a number of clinically important anticancer drugs, including carboplatin,92 docetaxel,93 topotecan,94 gefitinib,95 and erlotinib,96 and provided mathematical equations based on morphometric, demographic, phenotypic enzyme activity, and/or physiologic characteristics of patients, in order to predict drug clearance with an acceptable degree of precision and bias.97
REFERENCES
1. DeVita-Skin VT, Chu E. A history of cancer chemotherapy. Cancer Res 2008;68:8643–8653.
2. Lieu CH, Tan AC, Leong S, et al. From bench to bedside: lessons learned in translating preclinical studies in cancer drug development. J Natl Cancer Inst 2013;105:1441–1456.
3. Sparreboom A, Verweij J. Advances in cancer therapeutics. Clin Pharmacol Ther 2009;85:113–117.
4. Fujita KI, Sasaki Y. Optimization of cancer chemotherapy on the basis of pharmacokinetics and pharmacodynamics: from patients enrolled in ‘clinical trials’ to those in the ‘real world’. Drug Metab Pharmacokin 2014;29(1):20–28.
5. Liliemark J, Peterson C. Pharmacokinetic optimisation of anticancer therapy. Clin Pharmacokinet 1991;21:213–231.
6. Stuurman FE, Nuijen B, Beijnen JH, et al. Oral anticancer drugs: mechanisms of low bioavailability and strategies for improvement. Clin Pharmacokinet 2013;52:399–414.
7. Hasovits C, Clarke S. Pharmacokinetics and pharmacodynamics of intraperitoneal cancer chemotherapeutics. Clin Pharmacokinet 2012;51:203–224.
8. Cai S, Bagby TR, Forrest ML. Development of regional chemotherapies: feasibility, safety and efficacy in clinical use and preclinical studies. Ther Deliv 2011;2:1467–1484.
9. DeMario MD, Ratain MJ. Oral chemotherapy: rationale and future directions. J Clin Oncol 1998;16:2557–2567.
10. Zou P, Zheng N, Yang Y, et al. Prediction of volume of distribution at steady state in humans: comparison of different approaches. Exp Opin Drug Metab Toxicol 2012;8:855–872.
11. Deeken JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res 2007;13:1663–1674.
12. Pitz MW, Desai A, Grossman SA, et al. Tissue concentration of systemically administered antineoplastic agents in human brain tumors. J Neurooncol 2011;104:629–638.
13. Mruk DD, Su L, Cheng CY. Emerging role for drug transporters at the blood-testis barrier. Trends Pharmacol Sci 2011;32:99–106.
14. Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res 2000;17:1278–1283.
15. Malingre MM, Terwogt JM, Beijnen JH, et al. Phase I and pharmacokinetic study of oral paclitaxel. J Clin Oncol 2000;18:2468–2475.
16. Sparreboom A, Chen H, Acharya MR, et al. Effects of alpha1-acid glycoprotein on the clinical pharmacokinetics of 7-hydroxystaurosporine. Clin Cancer Res 2004;10:6840–6846.
17. Yamaoka K, Takakura Y. Analysis methods and recent advances in nonlinear pharmacokinetics from in vitro through in loci to in vivo. Drug Metab Dispos 2004;19:397–406.
18. van Zuylen L, Karlsson MO, Verweij J, et al. Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemother Pharmacol 2001;47:309–318.
19. Karlsson MO, Molnar V, Bergh J, et al. A general model for time-dissociated pharmacokinetic-pharmacodynamic relationship exemplified by paclitaxel myelosuppression. Clin Pharmacol Ther 1998;63:11–25.
20. Zhou Q, Gallo JM. The pharmacokinetic/pharmacodynamic pipeline: translating anticancer drug pharmacology to the clinic. AAPS J 2011;13:111–120.
21. Xie R, Mathijssen RH, Sparreboom A, et al. Clinical pharmacokinetics of irinotecan and its metabolites in relation with diarrhea. Clin Pharmacol Ther 2002;72:265–275.
22. Xie R, Mathijssen RH, Sparreboom A, et al. Clinical pharmacokinetics of irinotecan and its metabolites: a population analysis. J Clin Oncol 2002;20:3293–3301.
23. Kearns CM, Gianni L, Egorin MJ. Paclitaxel pharmacokinetics and pharmacodynamics. Sem Oncol 1995;22:16–23.
24. Minami H, Sasaki Y, Saijo N, et al. Indirect-response model for the time course of leukopenia with anticancer drugs. Clin Pharmacol Ther 1998;64:511–521.
25. Latz JE, Schneck KL, Nakagawa K, et al. Population pharmacokinetic/pharmacodynamic analyses of pemetrexed and neutropenia: effect of vitamin supplementation and differences between Japanese and Western patients. Clin Cancer Res 2009;15:346–354.
26. Karlsson MO, Anehall T, Friberg LE, et al. Pharmacokinetic/pharmacodynamic modelling in oncological drug development. Basic Clin Pharmacol Toxicol 2005;96:206–211.
27. Keizer RJ, Huitema AD, Schellens JH, et al. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010;49:493–507.
28. Slevin ML, Clark PI, Joel SP, et al. A randomized trial to evaluate the effect of schedule on the activity of etoposide in small-cell lung cancer. J Clin Oncol 1989;7:1333–1340.
29. Hainsworth JD. Extended-schedule oral etoposide in selected neoplasms and overview of administration and scheduling issues. Drugs 1999;58 Suppl 3:51–56.
30. Gelderblom H, Mross K, ten Tije AJ, et al. Comparative pharmacokinetics of unbound paclitaxel during 1- and 3-hour infusions. J Clin Oncol 2002;20:574–581.
31. Woodward EJ, Twelves C. Scheduling of taxanes: a review. Curr Clin Pharmacol 2010;5:226–231.
32. Soepenberg O, Sparreboom A, Verweij J. Clinical studies of camptothecin and derivatives. Alkaloid Chem Biol 2003;60:1–50.
33. Undevia SD, Gomez-Abuin G, Ratain MJ. Pharmacokinetic variability of anticancer agents. Nat Rev Cancer 2005;5:447–458.
34. Gurney H. Dose calculation of anticancer drugs: a review of the current practice and introduction of an alternative. J Clin Oncol 1996;14:2590–2611.
35. Baker SD, Verweij J, Rowinsky EK, et al. Role of body surface area in dosing of investigational anticancer agents in adults, 1991–2001. J Natl Cancer Inst 2002;94:1883–1888.
36. Mathijssen RH, Sparreboom A. Influence of lean body weight on anticancer drug clearance. Clin Pharmacol Ther 2009;85:23.
37. Sparreboom A, Wolff AC, Mathijssen RH, et al. Evaluation of alternate size descriptors for dose calculation of anticancer drugs in the obese. J Clin Oncol 2007;25:4707–4713.
38. Bartelink IH, Rademaker CM, Schobben AF, et al. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin Pharmacokinet 2006;45:1077–1097.
39. McLeod HL, Relling MV, Crom WR, et al. Disposition of antineoplastic agents in the very young child. Br J Cancer 1992;18:S23–S29.
40. Hutson JR, Weitzman S, Schechter T, et al. Pharmacokinetic and pharmacogenetic determinants and considerations in chemotherapy selection and dosing in infants. Exp Opin Drug Metab Toxicol 2012;8:709–722.
41. Veal GJ, Hartford CM, Stewart CF. Clinical pharmacology in the adolescent oncology patient. J Clin Oncol 2010;28:4790–4799.
42. Lichtman SM. Pharmacology of aging and cancer: how useful are pharmacokinetic tests? Interdiscip Top Gerontol 2013;38:104–123.
43. Relling MV, Crom WR, Pieper JA, et al. Hepatic drug clearance in children with leukemia: changes in clearance of model substrates during remission-induction therapy. Clin Pharmacol Ther 1987;41:651–660.
44. Evans WE, Rodman JH, Relling MV, et al. Differences in teniposide disposition and pharmacodynamics in patients with newly diagnosed and relapsed acute lymphocytic leukemia. J Pharmacol Exp Ther 1992;260:71–77.
45. Powis G, Harris RN, Basseches PJ, et al. Effects of advanced leukemia on hepatic drug-metabolizing activity in the mouse. Cancer Chemother Pharmacol 1986;16:43–49.
46. Charles KA, Rivory LP, Brown SL, et al. Transcriptional repression of hepatic cytochrome P450 3A4 gene in the presence of cancer. Clin Cancer Res 2006;12:7492–7497.
47. Moore MM, Chua W, Charles KA, et al. Inflammation and cancer: causes and consequences. Clin Pharmacol Ther 2010;87:504–508.
48. Albekairy A, Alkatheri A, Fujita S, et al. Cytochrome P450 3A4FNx011B as pharmacogenomic predictor of tacrolimus pharmacokinetics and clinical outcome in the liver transplant recipients. Saudi J Gastroenterol 2013;19:89–95.
49. Franke RM, Carducci MA, Rudek MA, et al. Castration-dependent pharmacokinetics of docetaxel in patients with prostate cancer. J Clin Oncol 2010;28:4562–4567.
50. Rahman A, White RM. Cytotoxic anticancer agents and renal impairment study: the challenge remains. J Clin Oncol 2006;24:533–536.
51. Egorin MJ, Van Echo DA, Olman EA, et al. Prospective validation of a pharmacologically based dosing scheme for the cis-diamminedichloroplatinum(II) analogue diamminecyclobutanedicarboxylatoplatinum. Cancer Res 1985;45:6502–6506.
52. Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol 1989;7:1748–1756.
53. Huang SM, Temple R, Xiao S, et al. When to conduct a renal impairment study during drug development: US Food and Drug Administration perspective. Clin Pharmacol Ther 2009;86:475–479.
54. Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther 2009;85:305–311.
55. Gibbons J, Egorin MJ, Ramanathan RK, et al. Phase I and pharmacokinetic study of imatinib mesylate in patients with advanced malignancies and varying degrees of renal dysfunction: a study by the National Cancer Institute Organ Dysfunction Working Group. J Clin Oncol2008;26:570–576.
56. Franke RM, Sparreboom A. Inhibition of imatinib transport by uremic toxins during renal failure. J Clin Oncol 2008;26:4226–4227.
57. Twelves C, Glynne-Jones R, Cassidy J, et al. Effect of hepatic dysfunction due to liver metastases on the pharmacokinetics of capecitabine and its metabolites. Clin Cancer Res 1999;5:1696–1702.
58. Eklund JW, Trifilio S, Mulcahy MF. Chemotherapy dosing in the setting of liver dysfunction. Oncology (Williston Park) 2005;19:1057–1063.
59. Hooker AC, Ten Tije AJ, Carducci MA, et al. Population pharmacokinetic model for docetaxel in patients with varying degrees of liver function: incorporating cytochrome P4503A activity measurements. Clin Pharmacol Ther 2008;84:111–118.
60. Grandison MK, Boudinot FD. Age-related changes in protein binding of drugs: implications for therapy. Clin Pharmacokinet 2000;38:271–290.
61. Sparreboom A, Nooter K, Loos WJ, et al. The (ir)relevance of plasma protein binding of anticancer drugs. Neth J Med 2001;59:196–207.
62. Perdaems N, Bachaud JM, Rouzaud P, et al. Relation between unbound plasma concentrations and toxicity in a prolonged oral etoposide schedule. Eur J Clin Pharmacol 1998;54:677–683.
63. Sparreboom A, van ZL, Brouwer E, et al. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res 1999;59:1454–1457.
64. Gardner ER, Burger H, van Schaik RH, et al. Association of enzyme and transporter genotypes with the pharmacokinetics of imatinib. Clin Pharmacol Ther 2006;80:192–201.
65. Wu K, House L, Ramirez J, et al. Evaluation of utility of pharmacokinetic studies in phase I trials of two oncology drugs. Clin Cancer Res 2013;19:6039–6043.
66. Hu S, Mathijssen RH, de Bruijn P, et al. Inhibition of OATP1B1 by tyrosine kinase inhibitors: in vitro-in vivo correlations. Br J Cancer 2014;110(4):894–898.
67. Kehrer DF, Mathijssen RH, Verweij J, et al. Modulation of irinotecan metabolism by ketoconazole. J Clin Oncol 2002;20:3122–3129.
68. van Leeuwen RW, Brundel DH, Neef C, et al. Prevalence of potential drug-drug interactions in cancer patients treated with oral anticancer drugs. Br J Cancer 2013;108:1071–1078.
69. Budha NR, Frymoyer A, Smelick GS, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther 2012;92:203–213.
70. Sparreboom A, Cox MC, Acharya MR, et al. Herbal remedies in the United States: potential adverse interactions with anticancer agents. J Clin Oncol 2004;22:2489–2503.
71. Dy GK, Bekele L, Hanson LJ, et al. Complementary and alternative medicine use by patients enrolled onto phase I clinical trials. J Clin Oncol 2004;22:4810–4815.
72. Goey AK, Mooiman KD, Beijnen JH, et al. Relevance of in vitro and clinical data for predicting CYP3A4-mediated herb-drug interactions in cancer patients. Cancer Treat Rev 2013;39:773–783.
73. Mathijssen RH, Verweij J, De Bruijn P, et al. Effects of St. John’s wort on irinotecan metabolism. J Natl Cancer Inst 2002;94:1247–1249.
74. Cox MC, Low J, Lee J, et al. Influence of garlic (Allium sativum) on the pharmacokinetics of docetaxel. Clin Cancer Res 2006;12:4636–4640.
75. van Erp NP, Baker SD, Zhao M, et al. Effect of milk thistle (Silybum marianum) on the pharmacokinetics of irinotecan. Clin Cancer Res 2005;11:7800–7806.
76. Wheeler HE, Maitland ML, Dolan ME, et al. Cancer pharmacogenomics: strategies and challenges. Nat Rev Genet 2013;14:23–34.
77. Fujita K, Sparreboom A. Pharmacogenetics of irinotecan disposition and toxicity: a review. Curr Clin Pharmacol 2010;5:209–217.
78. Huang RS, Ratain MJ. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J Clin 2009;59:42–55.
79. Evans WE, McLeod HL. Pharmacogenomics—drug disposition, drug targets, and side effects. N Engl J Med 2003;348:538–549.
80. Sparreboom A, Danesi R, Ando Y, et al. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat 2003;6:71–84.
81. Cusatis G, Gregorc V, Li J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst 2006;98:1739–1742.
82. Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest 2003;33 Suppl 2:1–5.
83. Smith NF, Figg WD, Sparreboom A. Role of the liver-specific transporters OATP1B1 and OATP1B3 in governing drug elimination. Exp Opin Drug Metab Toxicol 2005;1:429–445.
84. Sprowl JA, Mikkelsen TS, Giovinazzo H, et al. Contribution of tumoral and host solute carriers to clinical drug response. Drug Resist Updat 2012;15:5–20.
85. Kim DH, Sriharsha L, Xu W, et al. Clinical relevance of a pharmacogenetic approach using multiple candidate genes to predict response and resistance to imatinib therapy in chronic myeloid leukemia. Clin Cancer Res 2009;15:4750–4758.
86. Canal P, Chatelut E, Guichard S. Practical treatment guide for dose individualisation in cancer chemotherapy. Drugs 1998;56:1019–1038.
87. Evans WE, Relling MV, Rodman JH, et al. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med 1998;338:499–505.
88. Blasdel C, Egorin MJ, Lagattuta TF, et al. Therapeutic drug monitoring in CML patients on imatinib. Blood 2007;110:1699–1701.
89. Larson RA, Druker BJ, Guilhot F, et al. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood 2008;111:4022–4028.
90. Picard S, Titier K, Etienne G, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood 2007;109:3496–3499.
91. Blanchet B, Billemont B, Cramard J, et al. Validation of an HPLC-UV method for sorafenib determination in human plasma and application to cancer patients in routine clinical practice. J Pharm Biomed Anal 2009;49:1109–1114.
92. Chatelut E, Canal P, Brunner V, et al. Prediction of carboplatin clearance from standard morphological and biological patient characteristics. J Natl Cancer Inst 1995;87:573–580.
93. Bruno R, Hille D, Riva A, et al. Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J Clin Oncol 1998;16:187–196.
94. Gallo JM, Laub PB, Rowinsky EK, et al. Population pharmacokinetic model for topotecan derived from phase I clinical trials. J Clin Oncol 2000;18:2459–2467.
95. Li J, Karlsson MO, Brahmer J, et al. CYP3A phenotyping approach to predict systemic exposure to EGFR tyrosine kinase inhibitors. J Natl Cancer Inst 2006;98:1714–1723.
96. Lu JF, Eppler SM, Wolf J, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther 2006;80:136–145.
97. Mathijssen RH, de Jong FA, Loos WJ, et al. Flat-fixed dosing versus body surface area based dosing of anticancer drugs in adults: does it make a difference? Oncologist 2007;12:913–923.