Matthew P. Goetz, Charles Erlichman, Charles L. Loprinzi, and Manish Kohli
INTRODUCTION
Hormonal agents are commonly used as a treatment of hormonally responsive cancers, such as breast, prostate, or endometrial carcinomas. Other uses for some hormonal therapies include the treatment of paraneoplastic syndromes, such as carcinoid syndrome, and symptoms caused by cancer, including anorexia. This chapter discusses the major hormonal agents for such therapy, first with an overview of their use in practice, then with more detailed pharmacologic information regarding them (Table 27.1).

SELECTIVE ESTROGEN RECEPTOR MODULATORS
Tamoxifen
Tamoxifen continues to be an important hormonal therapy for the prevention and treatment of breast cancer worldwide. The continued importance of tamoxifen is reflected in the fact that it is the only hormonal agent approved by the U.S. Food and Drug Administration (FDA) for the prevention of premenopausal breast cancer,1 the treatment of ductal carcinoma in situ (DCIS),2 and the treatment of surgically resected premenopausal estrogen receptor (ER)–positive breast cancer.3
The standard daily dose of tamoxifen is 20 mg, and the optimal duration depends on the underlying clinical setting. Although the recommended duration in the prevention and DCIS settings is 5 years, recently published prospective studies have demonstrated that for the adjuvant treatment of invasive breast cancer, a duration of 10 years (compared to 5 years) further reduced the risk of breast cancer mortality and improved overall survival.4
The most common toxicity from tamoxifen is hot flashes, affecting approximately 50% of treated women. These hot flashes are of varying intensity and duration. Tamoxifen-induced hot flashes appear to increase over the first 3 months of therapy and then plateau. They appear to be more prominent in women with a history of hot flashes or estrogen replacement use. Tamoxifen-induced hot flashes can be ameliorated by a number of different pharmacotherapies, including low doses of megestrol5; antidepressants such as venlafaxine,6 desvenlafaxine,7 citalopram,8 escitalopram,9 and paroxetine10; and the anticonvulsant drugs gabapentin11 and pregabalin.12 There is evidence that drugs that inhibit CYP2D6 (e.g., paroxetine) alter the metabolic activation of tamoxifen to endoxifen, a critical metabolite associated with in vivo tamoxifen efficacy.13
The estrogenic properties of tamoxifen are responsible for both beneficial and deleterious side effects. Tamoxifen increases the incidence of endometrial cancer in postmenopausal (but not premenopausal) women, with the increase in the annual incidence of endometrial cancer being approximately 2.58 (ratio of incidence rates).14 The absolute risk depends on the duration of tamoxifen administration. For women who receive 10 years of adjuvant tamoxifen, the cumulative risk is 3.1% (mortality, 0.4%) versus 1.6% (mortality, 0.2%) for 5 years of tamoxifen.4 The incidence of a rarer form of uterine cancer, uterine sarcoma, is also increased after tamoxifen use.15 This form of endometrial cancer comprises approximately 15% of all uterine malignancies that develop after tamoxifen use.15 Beneficial estrogenic effects from tamoxifen include a decrease in total cholesterol16 and the preservation of bone density in postmenopausal women.17 In premenopausal women, however, tamoxifen has a negative effect on bone density.18 Although most patients do not complain of vaginal symptoms, a few complain of vaginal dryness, whereas others have increased vaginal secretions and discharge, the latter of which is an indication of the estrogenic activity of tamoxifen on the vagina. In the Arimidex, Tamoxifen, Alone or in Combination (ATAC) trial, a commonly observed tamoxifen side effect was vaginal bleeding, leading to a higher hysterectomy rate for patients randomized to tamoxifen (5%) compared to anastrozole (1%).19 An uncommon effect from tamoxifen is retinal toxicity. This drug can also increase the risk of cataracts. However, no difference in the rate of vision-threatening ocular toxicity has been seen among prospectively treated tamoxifen patients.20 Tamoxifen predisposes patients to thromboembolic phenomena, especially if used with concomitant chemotherapy. Depression has also been described, but the association with tamoxifen is not clear. Although liver cancers have been noted in laboratory animals, there is no established association between tamoxifen and liver cancers in humans.
Pharmacology
Tamoxifen acts by blocking estrogen stimulation of breast cancer cells, inhibiting both translocation and nuclear binding of the ER. This alters transcriptional and posttranscriptional events mediated by this receptor.21 Tamoxifen has agonistic, partial agonistic, or antagonistic effects depending on the species, tissue, or endpoints that have been assessed. Additionally, there are marked differences between the antiproliferative properties of tamoxifen and its metabolites.22
Resistance to tamoxifen can be intrinsic or acquired, and the potential mechanisms for this resistance are reviewed in the following paragraphs. At each step of the signal transduction pathway with which tamoxifen or its metabolites interferes, there is the potential for an alteration in response. The most important factor appears to be the level of ER, which is highly predictive for a response to tamoxifen. Tamoxifen is ineffective in ER-negative breast cancer. Although decreased or absent expression of the progesterone receptor (PR) is associated with a worse prognosis, the relative risk reduction in tamoxifen-treated patients is the same regardless of the presence or absence of the PR.
Following binding to the ER, subsequent translocation of the tamoxifen/ER complex to the nucleus and binding to an estrogen-response element may occur. This binding prevents transcriptional activation of estrogen-responsive genes. Laboratory and clinical data have demonstrated that ER-positive breast cancers that overexpress HER2 may be less responsive to tamoxifen and to hormonal therapy in general.23–26In these tumors, ligand-independent activation of the ER by mitogen-activated protein kinase (MAPK) pathways may contribute to resistance.27–29 In addition, the expression of AIB1, an estrogen-receptor coactivator, has been associated with tamoxifen resistance in patients whose breast cancers overexpress HER2.30 In some cases, resistance may result from a decrease or loss of ER expression.31,32 Although mutations in the ER ligand binding domain (LBD) are rare in newly diagnosed breast cancer, ER mutations are present in up to 20% of recurrent breast cancers.33–36 These mutations lead to a conformational change in the LBD, which mimics the conformation of activated ligand-bound receptor and constitutive, ligand-independent transcriptional activity, resulting in resistance to hormonal therapy. Preclinical studies suggest that some of these mutations, although insensitive to aromatase inhibitors, retain sensitivity to higher dose selective estrogen-receptor modulators (SERM), such as endoxifen, as well as fulvestrant.35
The carcinogenic potential of tamoxifen has been recognized in rat studies37–39 and in humans (endometrial cancer).40 It has been proposed that the generation of reactive intermediates that bind covalently to macromolecules underlies the process. Such reactive intermediates have been demonstrated in vitro.40–43 In addition, the induction of covalent DNA adducts in rat livers treated with tamoxifen has been reported.44 Both constitutive and inducible cytochrome P-450 (CYP) enzymes have been implicated in the formation of metabolites with tamoxifen,45,46 and the flavone-containing monooxygenase has been implicated in the formation of the N-oxide of tamoxifen. Reactive intermediates from such metabolic steps are being evaluated for their carcinogenic potential in vitro and in vivo.
Multiple studies to evaluate tumor gene expression profiling have identified gene expression patterns or specific genes associated with resistance to tamoxifen therapy. A commonly utilized gene expression assay, Oncotype DX 21 gene assay (Genomic Health, Redwood City, California), measures the expression of genes known to be involved in estrogen signaling (e.g., ER, PR), HER2, proliferation (e.g., Ki-67), and others. In multiple different data sets, the recurrence score has been associated with a higher risk of breast cancer recurrence in patients treated with hormonal therapy (e.g., tamoxifen or aromatase inhibitors) without concomitant chemotherapy.47–49
The pharmacokinetics of tamoxifen is complex. The chemical structure and metabolic pathway of tamoxifen are shown in Figure 27.1. Metabolic activation of tamoxifen is associated with greater pharmacologic activity. The two most active tamoxifen metabolites are 4-hydroxytamoxifen (4-OH tamoxifen) and 4-OH-N-desmethyltamoxifen (endoxifen). A series of studies carried out to characterize endoxifen pharmacology have demonstrated that it has equivalent potency in vitro to 4-hydroxytamoxifen in ER-α and -beta (ER-β) binding,50 for the suppression of ER-dependent human breast cancer cell line proliferation,22,50 and in global ER-responsive gene expression.51 A recent study suggests that endoxifen’s effect on the ER may differ from 4-hydroxytamoxifen based on the observation of ER-α degradation.52
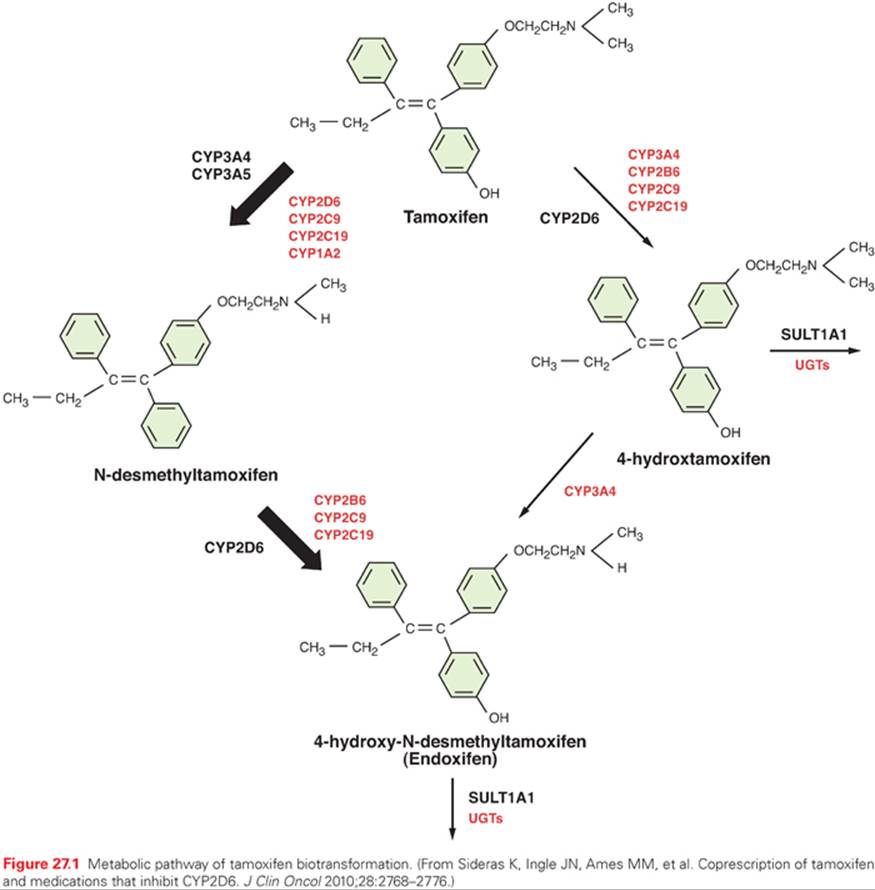
In women who receive tamoxifen at a dose of 20 mg per day, plasma endoxifen steady-state concentrations are generally 6 to 10 times higher than 4-hydroxytamoxifen.53 Although the metabolism of tamoxifen to 4-OH-tamoxifen is catalyzed by multiple enzymes, endoxifen is formed predominantly by the CYP2D6-mediated oxidation of N-desmethyltamoxifen, the most abundant tamoxifen metabolite (see Fig. 27.1).54 Multiple clinical studies have demonstrated that common CYP2D6 genetic variation (leading to low or absent CYP2D6 activity) or the drug-induced inhibition of CYP2D6 significantly lowers endoxifen concentrations.53,55 The CYP2D6 gene is highly polymorphic, with more than 70 major alleles with four well-defined phenotypes: poor metabolizers (PM), intermediate metabolizers (IM), extensive metabolizers (EM), and ultrarapid metabolizers (UM).
The clinical studies to evaluate the association between CYP2D6 polymorphisms and tamoxifen outcomes have yielded conflicting results. Initial56 and follow-up data57,58 demonstrated that CYP2D6 PM had an approximately two- to threefold higher risk of breast cancer recurrence (compared to CYP2D6 EM) and these data led an FDA special emphasis panel to recommend a tamoxifen label change to incorporate data that the CYP2D6 genotype was an important biomarker associated with tamoxifen efficacy.5 However, this label change has been delayed, in part because of conflicting data from secondary analyses of 5-year tamoxifen prospective trials (ATAC,59 BIG 1-98,60 and ABCSG861) as well as meta-analyses,62 which demonstrate that the CYP2D6 genotype is associated with tamoxifen efficacy when tamoxifen is administered as monotherapy for the adjuvant treatment of postmenopausal, ER-positive breast cancer. Additional support for the importance of endoxifen concentrations came from a secondary analysis of a prospective study, which demonstrated a higher risk of recurrence for women with low endoxifen concentrations.13
Many drugs are known to inhibit CYP2D6 activity. In tamoxifen-treated women, the coadministration of potent CYP2D6 inhibitors, such as paroxetine, converts a patient with normal CYP2D6 metabolism to a phenotypic PM.63Many other clinically important drugs have been reported to inhibit the CYP2D6 enzyme system, but their effects on tamoxifen metabolism have not been prospectively studied. As with the data regarding CYP2D6 genotype, the data regarding CYP2D6 inhibitors has additionally been controversial, including two studies that reported opposite findings with regard to CYP2D6 inhibitor use and breast cancer recurrence or death.64,65 Although the CYP2D6 data remain controversial, we conclude that until results from prospective adjuvant studies are available, women should be counseled regarding the potential impact of the CYP2D6 genotype on the effectiveness of adjuvant tamoxifen, and potent CYP2D6 inhibitors should be avoided. Additional caution should be used with drugs that induce CYP3A, such as rifampicin, as a these drugs have been demonstrated to substantially reduce (up to 86%) the concentrations of tamoxifen and its metabolites.66
Strategies to overcome low endoxifen concentrations include dose escalation of tamoxifen to 40 mg per day, which has been demonstrated to significantly increase endoxifen concentrations,67,68 as well as the direct administration of endoxifen itself. The latter strategy is ongoing in multiple different clinical trials, and early reports suggest clinical activity in aromatase inhibitors (AI)-resistant breast cancer.69
Following the metabolic activation of tamoxifen, the hydroxylated metabolites undergo both glucuronidation and sulfation. Peak plasma levels of tamoxifen (maximum concentration [Cmax]) are seen 3 to 7 hours after oral administration. Assuming an oral bioavailability of 30%, the volume of distribution has been calculated to be 20 L/kg, and plasma clearance ranges from 1.2 to 5.1 L per hour.70 The terminal half-life of tamoxifen has been reported to range between 4 and 11 days.71,72 The elimination half-life of tamoxifen increases with successive doses, which is consistent with saturable kinetics.71,73 The drug’s distribution in tissues is extensive. Levels of the parent drug and metabolites have been reported to be higher in tissue than in plasma in animal studies.74,75 Reports of tamoxifen concentrations 10- to 60-fold higher than plasma concentrations in the liver, lungs, brain, pancreas, skin, and bones are reported.76,77 Elevated levels of tamoxifen with biliary obstruction have been reported.78
Tamoxifen has been reported to interact with warfarin,73,79–81 digitoxin, phenytoin,82 and medroxyprogesterone.73 Tamoxifen-induced activation of human transcription factor pregnane X receptor (hPXR), resulting in the induction of CYP3A4, may increase the elimination of concomitantly administered CYP3A substrates,83 such as anastrozole.84
Toremifene
Toremifene is an agent similar to tamoxifen. It is available in the United States for the treatment of patients with metastatic breast cancer, and is approved in other countries for the adjuvant treatment of ER-positive breast cancer. Clinical trials have demonstrated no difference in either disease-free or overall survival when toremifene was compared with tamoxifen for the treatment of ER-positive breast cancer,85,86and evidence exists for major cross-resistance between tamoxifen and toremifene.87,88
Pharmacology
Toremifene is an antiestrogen with a chemical structure that differs from that of tamoxifen by the substitution of a chlorine for a hydrogen atom that is retained when toremifene undergoes metabolism.89 Like tamoxifen, toremifene is metabolized by CYP3A,90 with a secondary metabolism to form hydroxylated metabolites that appear to have similar binding affinities to 4-OH tamoxifen.89,91 The importance of these metabolites or the role of metabolism to the hydroxylated metabolites is unknown, but may play a role given the structural similarity of toremifene to tamoxifen. Although the oral bioavailability has not been defined, toremifene’s oral absorption appears to be good. The time to peak plasma concentrations after oral administration ranges from 1.5 to 6.0 hours,92 with the terminal half-lives for toremifene and one metabolite, 4-hydroxytoremifene, being 5 to 6 days.93,94 The apparent clearance is 5.1 L per hour. The terminal half-life for the major metabolite, N-desmethyltoremifene, is 21 days.95 The time to reach plasma steady-state concentrations is 1 to 5 weeks. Plasma protein binding is more than 99%. As with tamoxifen, toremifene is present at higher concentrations in tissues compared to plasma with a high apparent volume of distribution (958 L). Seventy percent of the drug is excreted in feces as metabolites. Studies in patients with impaired liver function or those on anticonvulsants known to induce CYP3A have demonstrated that hepatic dysfunction decreases the clearance of toremifene and N-desmethyltoremifene,95 whereas those patients on anticonvulsants had an increased clearance. Although toremifene appeared to be less carcinogenic than tamoxifen in preclinical models,43,96,97 of the rates of endometrial cancer in the adjuvant studies have been similar to tamoxifen.85
Raloxifene
Raloxifene is an estrogen agonist and antagonist originally developed to treat osteoporosis. Large placebo-controlled randomized trials demonstrated reduced rates of osteoporosis and a reduction in new breast cancers in treated women, leading to the development of a second-generation breast cancer chemoprevention trial (National Surgical Adjuvant Breast and Bowel Project, NSAPB P2) in which raloxifene was compared with tamoxifen in high-risk postmenopausal women. In this study, tamoxifen was superior to raloxifene in terms of both invasive and noninvasive cancer events, but was associated with a higher risk of thromboembolic events and endometrial cancer.98
Pharmacology
Raloxifene is partially estrogenic in bone99 and lowers cholesterol.100 It is antiestrogenic in mammary tissue101,102 and uterine tissue.103
The pharmacokinetics of raloxifene have been studied principally in postmenopausal women.104–106 Pharmacokinetic parameters of raloxifene show considerable interindividual variation. Limited information is available on the pharmacokinetics of raloxifene in individuals with hepatic impairment, renal impairment, or both.
Raloxifene is rapidly absorbed from the gastrointestinal tract. Because raloxifene undergoes extensive first-pass glucuronidation, oral bioavailability of unchanged drug is low. Although approximately 60% of an oral dose is absorbed, the absolute bioavailability as unchanged raloxifene is only 2%. However, systemic availability of raloxifene may be greater than that indicated in bioavailability studies, because circulating glucuronide conjugates are converted back to the parent drug in various tissues.
After the oral administration of a single 120- or 150-mg dose of raloxifene hydrochloride, peak plasma concentrations of raloxifene and its glucuronide conjugates are achieved at 6 hours and 1 hour, respectively. After the oral administration of radiolabeled raloxifene, less than 1% of total circulating radiolabeled material in plasma represents the parent drug.
Results of a single-dose study in patients with liver dysfunction indicate that plasma raloxifene concentrations correlate with serum bilirubin concentrations and are 2.5 times higher than individuals with normal hepatic function. In postmenopausal women who received raloxifene in clinical trials, plasma concentrations of raloxifene and the glucuronide conjugates in those with renal impairment (i.e., estimated creatinine clearance values as low as 23 mL per minute) were similar to values in women with normal renal function.
Raloxifene and its monoglucuronide conjugates are more than 95% bound to plasma proteins. Raloxifene binds to albumin and α1-acid glycoprotein. Raloxifene undergoes extensive first-pass metabolism to the glucuronide conjugates raloxifene 4′-glucuronide, 6-glucuronide, and 6,4′-diglucuronide. UGT1A1 and -1A8 have been found to catalyze the formation of both the 6-β-and 4′-β-glucuronides, whereas UGT1A10 formed only the 4′-β-glucuronide.107 The metabolism of raloxifene does not appear to be mediated by CYP enzymes (such as CYP2D6), because metabolites other than glucuronide conjugates have not been identified.
The plasma elimination half-life of raloxifene at steady state averages 32.5 hours (range, 15.8 to 86.6 hours). Raloxifene is excreted principally in feces as an unabsorbed drug and via biliary elimination as glucuronide conjugates, which, subsequently, are metabolized by bacteria in the gastrointestinal tract to the parent drug. After oral administration, less than 0.2% of a raloxifene dose is excreted as the parent compound and less than 6% as glucuronide conjugates in urine.
Fulvestrant
Fulvestrant is an ER antagonist that has no known agonist activity and results in ER downregulation.108–111 Like tamoxifen, fulvestrant competitively binds to the ER but with a higher affinity—approximately 100 times greater than that of tamoxifen,108,112–114—thus preventing endogenous estrogen from exerting its effect in target cells.
Results from two phase III clinical trials using the 250 mg per month dose demonstrated fulvestrant to be as effective as anastrozole in the treatment of postmenopausal women with advanced hormone receptor–positive breast cancer previously treated with antiestrogen therapy (mainly tamoxifen).112–116 In the setting of first-line hormone-responsive metastatic breast cancer, a randomized phase III clinical trial to compare tamoxifen to fulvestrant (250 mg per month) demonstrated no differences in response or time to progression.117 Because of pharmacology data (discussed in the following paragraphs), the 500 mg per day dose was developed. A randomized trial comparing the 250 mg per month with 500 mg per month dose demonstrated a 4-month improvement in median overall survival advantage for the higher dose.118 For this reason, the higher dose is now the standard recommended dose.
Fulvestrant is well tolerated. The most common drug-related events (greater than 10% incidence) from the randomized phase III studies were injection-site reactions and hot flashes. Common events (1% to 10% incidence) included asthenia, headache, and gastrointestinal disturbances such as nausea, vomiting, and diarrhea, with minor gastrointestinal disturbances being the most commonly described adverse event.
Pharmacology
Fulvestrant is a steroidal molecule derived from E2 with an alkylsulphonyl side chain in the 7-α position (Fig. 27.2). Because fulvestrant is poorly soluble and has low and unpredictable oral bioavailability, a parenteral formulation of fulvestrant was developed in an attempt to maximize delivery of the drug.111 The intramuscular formulation provides prolonged release of the drug over several weeks. The pharmacokinetics of three different single doses of fulvestrant (50, 125, and 250 mg) have been published.111 In this phase I/II multicenter study, postmenopausal women with primary breast cancer who were awaiting curative surgery received either fulvestrant, tamoxifen, or placebo. After single intramuscular injections of fulvestrant, the time of maximal concentration (tmax) ranged from 2 to 19 days, with the median being 7 days for each dose group. At the interval of 28 days, Cmin values were two- to fivefold lower than the Cmax values. For most patients in the 125- and 250-mg dose groups, significant levels of fulvestrant were still measurable 84 days after administration. Pharmacokinetic modeling of the pooled data from the 250-mg cohort was best described by a two-compartment model in which a longer terminal phase began approximately 3 weeks after administration. Because of the long time needed to reach a steady state, the 500-mg loading dose regimen was prospectively studied and determined to be superior to the 250 mg per month dose, both in terms of steady state concentrations achieved within 1 month119as well as progression-free and overall survival.118

AROMATASE INHIBITORS
At menopause, the synthesis of ovarian hormones ceases. However, estrogen continues to be converted from androgens (produced by the adrenal glands) by aromatase, an enzyme of the CYP superfamily. Aromatase is the enzyme complex responsible for the final step in estrogen synthesis via the conversion of androgens, androstenedione and testosterone, to estrogens, estrone (E1) and E2. This biologic pathway served as the basis for the development of the antiaromatase class of compounds. Alterations in aromatase expression have been implicated in the pathogenesis of estrogen-dependent disease, including breast cancer, endometrial cancer, and endometriosis. The importance of this enzyme is also highlighted by the fact that selective aromatase inhibitors are commonly used as first-line therapy for the treatment of postmenopausal women with estrogen-responsive breast cancer. Aminoglutethimide was the first clinically used aromatase inhibitor. When it became available, it was used to cause a medical adrenalectomy. Because of the lack of selectivity for aromatase and the resultant suppression of aldosterone and cortisol, aminoglutethimide is no longer recommended for treating metastatic breast cancer. Aminoglutethimide is also occasionally used to try to reverse excess hormone production by adrenocortical cancers.120
Aromatase (cytochrome P-450 19 [CYP19]) is encoded by the CYP19 gene, which is highly polymorphic. Some of these variants are functionally important121 and may have clinical significance.122,123
Aromatase inhibitors have been classified in a number of different ways, including first, second, and third generation; steroidal and nonsteroidal; and reversible (ionic binding) and irreversible (suicide inhibitor, covalent binding).124 The nonsteroidal aromatase inhibitors include aminoglutethimide (first generation), rogletimide and fadrozole (second generation), and anastrozole, letrozole, and vorozole (third generation). The steroidal aromatase inhibitors include formestane (second generation) and exemestane (third generation).
Steroidal and nonsteroidal aromatase inhibitors differ in their modes of interaction with, and their inactivation of, the aromatase enzyme. Steroidal inhibitors compete with the endogenous substrates, androstenedione and testosterone, for the active site of the enzyme and are processed into intermediates that bind irreversibly to the active site, causing irreversible enzyme inhibition.19 Nonsteroidal inhibitors also compete with the endogenous substrates for access to the active site, where they then form a reversible bond to the heme iron atom so that enzyme activity can recover if the inhibitor is removed; however, inhibition is sustained whenever the inhibitor is present.19
Letrozole and Anastrozole
Both letrozole and anastrozole have been extensively studied in the metastatic and adjuvant settings. When compared to tamoxifen, both letrozole and anastrozole have demonstrated superior response rates and progression-free survival in the metastatic setting.124,125 In the adjuvant setting, two trials have been performed and demonstrated superiority in terms of relapse-free survivals of both anastrozole (ATAC)126and letrozole (BIG 1-98).127 Additionally, anastrozole has been studied in a sequential approach, and the sequence of tamoxifen followed by anastrozole is superior to 5 years of tamoxifen alone.128 Anastrozole has recently been compared to placebo in women at an increased risk of developing breast cancer and was demonstrated to significantly reduce the incidence of invasive breast cancer.129
The side effects of both anastrozole and letrozole are similar and include arthralgias and myalgias in up to 50% of patients. Both letrozole and anastrozole are associated with a higher rate of bone fracture, compared with the tamoxifen.130 At the present time, minimal long-term (longer than 5 years) clinical data regarding the effect of aromatase inhibitors on bones are available. When offering anastrozole for extended periods of time to patients with early breast cancer, attention to bone health is paramount, and bone density should be monitored in all patients. Prospective studies have demonstrated that bisphosphonates prevent aromatase-inhibitor–induced bone loss and a meta-analysis presented at the 2013 San Antonio Breast Cancer Symposium demonstrated that bisphosphonates reduce bone recurrences and prolong overall survival. Therefore, bisphosphonates should be considered in AI-treated patients, both in those with and without an increased risk of bone fractures.
A meta-analysis of toxicities comparing aromatase inhibitors with tamoxifen has demonstrated a 30% increase in grade 3 and 4 cardiac events with aromatase inhibitors.131 However, prospective data demonstrate no differences in myocardial events comparing anastrozole with placebo, although an increase in hypertension was observed.129
No impact has been seen with anastrozole on adrenal steroidogenesis at up to 10 times the clinically recommended dose.132 Although letrozole may decrease basal and adrenocorticotropic hormone–stimulated cortisol synthesis,133,134 the clinical effect appears to be minimal. Aromatase inhibitors appear to have differential effects on lipids. In a study of over 900 patients with metastatic disease, anastrozole showed no marked effect on lipid profiles compared with baseline.135 Conversely, the administration of letrozole in women with advanced breast cancer resulted in significant increases in total cholesterol and low-density lipoprotein, from baseline, after 8 and 16 weeks of therapy.136 In the Breast International Group 1-98 trial, more women who received letrozole experienced grade 1 hypercholesterolemia compared to women who received tamoxifen.127
Letrozole is a nonsteroidal aromatase inhibitor with a high specificity for the inhibition of estrogen production (Fig. 27.3). Letrozole is 180 times more potent than aminoglutethimide as an inhibitor of aromatase in vitro. Aldosterone production in vitro is inhibited by concentrations 10,000 times higher than those required for inhibition of estrogen synthesis.137,138 In a normal male volunteer study, letrozole was shown to decrease E2 and serum E1levels to 10% of baseline with a single 3-mg dose. In phase I studies, letrozole caused a significant decline in plasma E1 and E2 within 24 hours of a single oral dose of 0.1 mg.139,140 After 2 weeks of treatment, the blood levels of E2, E1, and estrone sulfate were suppressed 95% or more from baseline. This continued over the 12 weeks of therapy. There was no apparent alteration in plasma levels of cortisol and aldosterone with letrozole or after corticotropin stimulation.139 In postmenopausal women with advanced breast cancer, the drug did not have any effect on follicle-stimulating hormone (FSH), luteinizing hormone (LH), thyrotropin (previously thyroid-stimulating hormone), cortisol, 17-α-hydroxyprogesterone, androstenedione, or aldosterone blood concentrations.141,142

Anastrozole is a nonsteroidal aromatase inhibitor that is 200-fold more potent than aminoglutethimide.143 No effect on the adrenal glands has been detected. In human studies, the tmax is 2 to 3 hours after oral ingestion.144Elimination is primarily via hepatic metabolism, with 85% excreted by that route and only 10% excreted unchanged in urine. The main circulating metabolite is triazole after cleavage of the two rings in anastrozole by N-dealkylation. Linear pharmacokinetics have been observed in the dose range of 1 to 20 mg and do not change with repeat dosing. The terminal half-life is approximately 50 hours, and steady-state concentrations are achieved in approximately 10 days with once-a-day dosing and are three to four times higher than peak concentrations after a single dose. Plasma protein binding is approximately 40%.145 In one study, anastrozole 1 mg and 10 mg daily, inhibited in vivo aromatization by 96.7% and 98.1%, respectively, and plasma E1 and E2 levels were suppressed 86.5% and 83.5%, respectively, regardless of dose.146 Thus, 1 mg of anastrozole achieves near maximal aromatase inhibition and plasma estrogen suppression in breast cancer patients.
A recent prospective study to evaluate the pharmacokinetics of anastrozole (1 mg per day) demonstrated large interindividual variations in plasma anastrozole and anastrozole metabolite concentrations, as well as pretreatment and postdrug plasma E1, E2, and E1 conjugate and estrogen precursor (androstenedione and testosterone) concentrations.147 Further research is needed to determine the basis for the wide variability in the pharmacokinetics of anastrozole and whether these findings are clinically relevant.
Exemestane
Exemestane has a steroidal structure and is classified as a type 1 aromatase inhibitor, also known as an aromatase inactivator, because it irreversibly binds with and permanently inactivates the enzyme.134Exemestane has been compared to tamoxifen in both the metastatic and adjuvant settings. In the setting of tamoxifen-refractory metastatic breast cancer, exemestane is superior to megestrol acetate, as demonstrated in a phase III trial in which improvements in both median time to tumor progression and median survival were observed.148 In the adjuvant setting, the international exemestane study compared 2 to 3 years of tamoxifen with 2 to 3 years of exemestane in women who had previously competed 2 to 3 years of adjuvant tamoxifen. In this trial, a switch to exemestane resulted in superior disease-free and overall survival in the hormone receptor–positive subtype. Furthermore, exemestane has been compared with the nonsteroidal agent anastrozole in the adjuvant treatment of ER-positive breast cancer, and there were no differences in disease-free or overall survival.149 Finally, exemestane has been compared to placebo in patients at increased risk of breast cancer, and a significant reduction in the risk of developing invasive breast cancer was observed.150
Side Effects of Exemestane
Although preclinical studies have suggested that exemestane prevented bone loss in ovariectomized rats,151 the Intergroup Exemestane adjuvant trial still demonstrated a higher rate of bone fracture for patients randomized to the exemestane arm and there were no differences in fracture rates comparing anastrozole with exemestane.149 Side effects, including arthralgias and myalgias, appear to be similar to the other AIs. With regard to steroidogenesis, no impact on either cortisol or aldosterone levels was seen in a small study after the administration of exemestane for 7 days.152 Finally, exemestane has weak androgenic properties, and its use at higher doses has been associated with steroidal-like side effects, such as weight gain and acne.153,154 However, these side effects have not been observed with the FDA-approved dose (25 mg per day).155
Pharmacology
Exemestane is administered once daily by mouth, with the recommended daily dose being 25 mg. The time needed to reach maximal E2 suppression is 7 days,156 and its half-life is 27 hours.157 At daily doses of 10 to 25 mg, exemestane suppresses estrogen concentrations to 6% to 15% of pretreatment levels. This activity is more pronounced than that produced by formestane and comparable to that produced by the nonsteroidal AIs, anastrozole and letrozole.158–160 Exemestane does not appear to affect cortisol or aldosterone levels when evaluated after 7 days of treatment based on dose-ranging studies, including doses from 0.5 to 800 mg.152 Exemestane is metabolized by CYP3A4.134 Although drug–drug interactions have not been formally reported for exemestane, there is the potential for interactions with drugs that affect CYP3A4.134
GONADOTROPIN-RELEASING HORMONE ANALOGS
Gonadotropin-releasing hormone (GnRH) analogs result in a medical orchiectomy in men and are used as a means of providing androgen ablation for hormone-sensitive and castration refractory metastatic prostate cancer.161 Because the initial agonist activity of GnRH analogs can cause a tumor flare from temporarily increased androgen levels, concomitant use of the antiandrogen flutamide or bicalutamide has been used to prevent this effect. GnRH analogs can also cause tumor regressions in hormonally responsive breast cancers162 and have received FDA approval for the treatment of metastatic breast cancer in premenopausal women. Data suggest that these drugs may be useful as adjuvant therapy of premenopausal women with resected breast cancer.163 The use of these drugs in combination with tamoxifen or exemestane in premenopausal women with primary breast cancer is the subject of large, ongoing, international clinical trials. The primary toxicities of GnRH analogs are secondary to the ablation of sex steroid concentrations and include hot flashes, sweating, and nausea.164 These symptoms can be reversed with low doses of progesterone analogs.5 In males treated with GnRH analogs for prostate cancer, an alternate strategy of intermittent schedule of GnRH administration may result in improved tolerability and quality of life, with comparable efficacy compared with continuous GnRH analog administration in well-selected advanced prostate cancer patient cohorts.165 However, in a recent trial comparing intermittent with continuous androgen ablation in newly diagnosed metastatic hormone sensitive prostate cancer patients, a greater risk for death from an intermittent strategy could not be conclusively ruled out although intermittent therapy resulted in small improvements in quality of life.166
GnRH analogs available for clinical use include goserelin167,168 and leuprolide.169 Both are available in depot intramuscular preparations to be given at monthly intervals. The recommended monthly dose of leuprolide is 7.5 mg and of goserelin is 3.6 mg. There are also longer acting depot preparations to be administered every 3, 4, 6, and 12 months.
Pharmacology
Analogs of the decapeptide GnRH167,169,170 have been synthesized by modifications of position 6 in which the l-glycine has been exchanged for a d-amino acid and the C-terminal amino acid has been either replaced by an ethylamide or substituted for a modified amino acid. These changes increase the affinity of the analog for the GnRH receptor and decrease the susceptibility to enzymatic degradation. There is an amino acid structure of GnRH with the substitutions for leuprolide and goserelin. Initial administration of these compounds results in stimulation of gonadotropin release. However, prolonged administration has led to profound inhibition of the pituitary–gonadal axis.170Plasma E2 and progesterone are consistently suppressed to postmenopausal or castrate levels after 2 to 4 weeks of treatment with goserelin or leuprolide.164,171 These drugs are administered intramuscularly or subcutaneously in a parenteral sustained-release microcapsule preparation, because parenteral administration of the parent drug is otherwise associated with rapid clearance. The GnRH analogs are metabolized in the liver, kidney, hypothalamus, and pituitary gland by neutral peptidase cleavage of the peptide bond between the tyrosine in the 5 position and the amino acid in position 6 and by a postproline-cleaving enzyme that cleaves the peptide bond between proline in the 9 position and the glycine-NH2 in the 10 position. Substitutions at the glycine 6 position and modification of the C-terminal make these analogs more resistant to this enzymatic cleavage.
Leuprolide is approximately 80 to 100 times more potent than endogenous GnRH. It induces castrate levels of testosterone in men with prostate cancer within 3 to 4 weeks of drug administration after an initial sharp increase in LH and FSH. The mechanisms of action include pituitary desensitization after a reduction in pituitary GnRH receptor binding sites and possibly a direct antitumor effect in ER-positive human breast cancer cells.169 The depot form results in a dose rate of 210 μg per day of leuprolide. Peak concentrations of the depot form, achieved approximately 3 hours after drug administration, have been reported to range between 13.1 and 54.5 μg/L. There appears to be a linear increase in the area under the curve (AUC) for doses of 3.75, 7.5, and 15.0 mg in the depot form. The parenteral bioavailability of subcutaneously injected leuprolide is 94%. The volume of distribution ranges from 27.4 to 37.1 L. In human studies, leuprolide urinary excretion as a metabolite was the primary route of clearance.
Goserelin is approximately 100 times more potent than the naturally occurring GnRH. Like leuprolide, it causes the stimulation of LH and FSH acutely, and with subsequent administration, GnRH receptor numbers decrease, and the pituitary becomes desensitized with decreasing LH and FSH levels. Castrate levels of testosterone are achieved within 1 month. In women, goserelin inhibits ovarian androgen production, but serum levels of dehydroepiandrosterone sulfate and, to a lesser extent, androstenedione, are preserved. In vitro, goserelin has demonstrated antitumor activity in estrogen-dependent MCF7 human breast cancer cells and LNCaP2 prostate cancer cells. The drug is released at a continuous mean rate of 120 μg per day in the depot form, with peak concentrations in the range of 2 to 3 μg/L achieved. The mean volume of distribution in six patients has been reported to be 13.7 L,172 which is consistent with extracellular fluid volume. Goserelin is principally excreted in the urine, with a mean total body clearance of 8 L per hour in patients with normal renal function. The total body clearance is reduced by approximately 75%, with renal dysfunction and the elimination half-life increased two- or threefold. However, dose adjustment for renal insufficiency does not appear to be necessary. The 5 to 10 hexapeptide and the 4 to 10 hexapeptide were detected in urine in animal studies.173 The terminal half-life of goserelin is approximately 5 hours after subcutaneous injection. Protein binding is low, and no known drug interactions have been documented.
GONADOTROPIN-RELEASING HORMONE ANTAGONISTS
Modification to the structure of GnRH has resulted in the development of GnRH antagonist compounds that are currently being used in the treatment of prostate cancer. Abarelix was initially approved by the FDA in 2003 as the first depot-injectable GnRH antagonist, but was subsequently withdrawn in 2005. Degarelix is a synthetically modified compound with GnRH antagonist activity that was approved for use by the FDA in 2008 for the management of prostate cancer.174 Its effect in prostate cancer treatment is to block the GnRH receptor, and thereby prevent the trigger for the production of LH, which mediates androgen synthesis. In contrast to GnRH analogs, degarelix does not cause tumor flare symptoms secondary to temporary increased androgen production. A large randomized clinical trial demonstrated that degarelix was associated with a rapid and sustained reduction in serum testosterone, prostate-specific antigen (PSA), FSH, and LH levels, with a loading dose of 240 mg subcutaneously, followed by a monthly maintenance dose of 80 mg175 with comparable efficacy to leuprolide.176 The most common side effects (greater than 10%) were hot flashes and pain at the injection site176 when patients were provided degarelix for a 12-month period. It is unknown if degarelix will have a similar chronic side effect profile known to be associated with long-term GnRH analog use.
Pharmacology
The recommended loading dose of degarelix is 240 mg, administered as two injections of 120 mg each subcutaneously. Monthly maintenance doses of 80 mg as a 20 mg/mL solution is started 28 days after the loading dose. In an analysis of pharmacokinetic/pharmacodynamic (PK/PD) properties of degarelix in 60 healthy males, after a single subcutaneous dose, a terminal half life of 47 days was observed.177 PK properties of degarelix have been evaluated when administered as a subcutaneous depot of drug as a gel in six different doses to 48 healthy males and when administered intravenously. Using data from several clinical trials, the rate of drug diffusion from subcutaneous administration results in detectable drug up to 60 days after a single dose compared to less than 4 days when the drug is injected intravenously.
ANTIANDROGENS
Flutamide
The antiandrogen flutamide is used in men with metastatic prostate cancer either as initial therapy, combined with GnRH analog administration, or when the metastatic prostate cancer is unresponsive, despite androgen ablation therapy. The recommended dose is 250 mg by mouth three times a day. In patients whose prostate cancer is growing despite flutamide use, stopping flutamide can sometimes cause a flutamide-withdrawal response.
The most common toxicity seen with flutamide is diarrhea, with or without abdominal discomfort. Gynecomastia, which can be tender, frequently occurs in men who are not receiving concomitant androgen ablation therapy.178Flutamide can rarely cause hepatotoxicity, a condition that is reversible if detected early, but this toxicity can also be fatal.179 There is no accepted, clinically recommended testing schedule to screen for flutamide-induced hepatotoxicity other than being aware of this phenomenon and testing for liver function if hepatic symptoms develop.
Pharmacology
Flutamide is a pure antiandrogen with no intrinsic steroidal activity.180 Flutamide’s mechanism of action is as an androgen-receptor antagonist. This binding prevents dihydrotestosterone binding and subsequent translocation of the androgen-receptor complex into the nuclei of cells. Because it is a pure antiandrogen, it acts only at the cellular level. The administration of flutamide alone leads to increased LH and FSH production and a concomitant increase in plasma testosterone and E2 levels. Plasma protein binding ranges between 94% and 96% for flutamide and between 92% and 94% for 2-hydroxyflutamide, its major metabolite. When the drug is administered three times a day, steady state levels are achieved by day 6. The elimination half-life at steady state is 7.8 hours, and 2-hydroxyflutamide achieves concentrations 50 times higher than the parent drug at steady state and has equal or greater potency than that of flutamide.180 The elimination half-life for the metabolite is 9.6 hours. The high plasma concentrations of 2-hydroxyflutamide, as compared with flutamide, suggest that the therapeutic benefits of flutamide are mediated primarily through its active metabolite.181
Bicalutamide
Bicalutamide is another nonsteroidal antiandrogen that has been approved by the FDA for use in the United States. The recommended dose is one 50-mg tablet per day. One randomized trial reported that bicalutamide compared favorably with flutamide in patients with advanced prostate cancer.182 Bicalutamide appears to be relatively well tolerated and is associated with a lower incidence of diarrhea than is flutamide.
Pharmacology
Bicalutamide has a binding affinity to the androgen receptor in the rat prostate that is four times greater than that of 2-hydroxyflutamide.183,184 In vivo, bicalutamide caused a marked inhibition of growth of accessory sex organs in rats, with a potency 5 to 10 times greater than that of flutamide. Unlike flutamide, bicalutamide did not cause a significant increase in LH or testosterone in rats. In humans, the drug has a long plasma half-life of 5 to 7 days, so it may be administered on a weekly schedule. Pharmacokinetics of the drug showed a dose-dependent increase in mean peak plasma concentrations, and the AUC increased linearly with the dose. The half-life of bicalutamide in humans was approximately 6 days, and the drug clearance was not saturable at plasma concentrations up to 1,000 ng/mL. Daily dosing of the drug led to an approximately tenfold accumulation after 12 weeks of administration. In contrast to results in rats, serum concentrations of testosterone and LH increased significantly from baseline at all dose levels tested in humans. Whereas serum FSH concentrations remained essentially unchanged, the median serum E2concentrations increased significantly.185
Nilutamide
Nilutamide represents the third variation of an antiandrogen available for use in patients with prostate cancer. The observation of unique toxicities, night blindness, and pulmonary toxicity has limited its use.
NOVEL ANTIANDROGENS
Although testosterone depletion remains an unchallenged standard for advanced stage hormone-sensitive disease, evidence has emerged that castration-recurrent prostate cancer remains androgen receptor (AR) dependent and is neither hormone refractory nor androgen independent, which were commonly used terms to define the progression of advanced stage disease following androgen deprivation therapy. Recognition of AR functioning despite the paucity of circulating androgens is evidenced by the elevation of AR messenger RNA in castration-recurrent tumor tissue relative to androgen-dependent tumors and reexpression of some androgen-regulated genes during clinical castration resistance. Recently, the AR axis has been the focus of therapeutic targeting.
Abiraterone Acetate
After the failure of initial androgen manipulation with GnRH analogs and peripheral antiandrogens, prostate cancer continues to respond to a variety of second- and third-line hormonal interventions. Based on this observation, CYP17, a key enzyme in androgen and estrogen synthesis, was targeted using ketoconazole, which is a weak, reversible, and nonspecific inhibitor of CYP17 resulting in modest antitumor activity of short durability. More recently, abiraterone, a more potent (i.e., 20 times more than ketoconazole), selective, and irreversible inhibitor of CYP17, has been investigated in castration-recurrent prostate cancer, and significant objective responses have been observed.186 Chemically, it is a 3-pyridyl steroid pregnenolone–derived compound available in an oral prodrug form of abiraterone acetate. Its main toxicity is from symptoms of mineralocorticoid excess (including hypokalemia, hypertension, and fluid overload), because continuous CYP17 blockade results in raising adrenocorticotrophic hormone (ACTH) levels that increase steroid levels upstream of CYP17, including corticosterone and deoxycorticosterone. These adverse effects are best avoided by the coadministration of steroids.
The established dose of abiraterone is 1,000 mg a day (four 250 mg tablets). Following oral administration of abiraterone acetate, the median time to maximum plasma abiraterone concentrations is 2 hours. At the dose of 1,000 mg daily, steady state values (mean ± standard deviation [SD]) of Cmax were 226 ± 178 ng/mL and of AUC were 1173 ± 690 ng.hr/mL. Abiraterone is highly bound (>99%) to the human plasma proteins, albumin and alpha-1 acid glycoprotein. The apparent steady state volume of distribution (mean ± SD) is 19,669 ± 13,358 L. No major deviation from dose proportionality was observed in the dose range of 250 mg to 1,000 mg. However, the exposure was not significantly increased when the dose was doubled from 1,000 to 2,000 mg (8% increase in mean AUC). The two main circulating metabolites of abiraterone in human plasma are abiraterone sulfate (inactive) and N-oxide abiraterone sulfate (inactive), which each account for about 43% of exposure. CYP3A4 and SULT2A1 are enzymes involved in the formation and conjugation of N-oxide abiraterone.
Enzalutamide
Enzalutamide is a new diarylthiohydantoin compound that binds AR with an affinity that is several-fold greater than the antiandrogens bicalutamide and flutamide. This class of novel AR inhibitor also disrupts the nuclear translocation of AR and impairs DNA binding to androgen response elements and the recruitment of coactivators.187 In early clinical trials, promising results have been observed in castrate refractory and chemotherapy-resistant settings. The major metabolite of enzalutamide is N-desmethyl enzalutamide, and CYP2C8 is responsible for the formation of the active metabolite, N-desmethyl enzalutamide. Enzalutamide pharmacokinetics, in the studied dose range between 30 mg to 480 mg, exhibited a linear, two-compartmental model with first-order kinetics. In patients with mCRPC, the mean (% coefficient of variation [CV]) predose Cmin values for enzalutamide and N-desmethyl enzalutamide were 11.4 (25.9%) μg/mL and 13.0 (29.9%) μg/mL, respectively. Enzalutamide is mainly metabolized by CYP2C8 and CYP3A4. Doses ranging from 30 to 600 mg daily have been evaluated, with dose-limiting toxicities including fatigue, seizure, asthenia, anemia, and arthralgia occurring at higher dose levels. At present, enzalutamide has been approved for treating advanced castrate-recurrent prostate cancer188 after a failure of docetaxel chemotherapy at a dose of 160 mg (four, 40 mg oral capsules). Clinical trials are ongoing to evaluate the efficacy of enzalutamide in castrate-recurrent patients who are chemotherapy naïve.
Galeterone and Orteronel
Novel CYP17 inhibitors that are more selective for 17,20-lyase over 17 α-hydroxylase are currently being developed. Orteronel (TAK-700) is an example of a highly selective 17,20 lyase, which is currently undergoing phase III clinical trials in a pre- and postchemotherapy castrate-recurrent setting after the failure of androgen-deprivation therapy.189 Other novel agents being developed include galeterone, which is an inhibitor of CYP 17 α-hydroxylase and C17,20 lyase. Survival mechanisms of prostate cancer cells targeted by galeterone include its binding to AR, competitive inhibition of testosterone binding, and a reduction in the quantity of AR protein within the prostate cancer cells. It can also enhance the degradation of constitutively active splice variants. Therefore, taken together, it diminishes the ability of the cells to respond to the low levels of androgenic growth signals. This agent is currently in early clinical safety and efficacy testing for advanced stage prostate cancer.
OTHER SEX STEROID THERAPIES
Fluoxymesterone
Fluoxymesterone is an androgen that has been used in women with metastatic breast cancer who have hormonally responsive cancers and who have progressed on other hormonal therapies such as tamoxifen, an aromatase inhibitor, or megestrol acetate. The usual dose is 10 mg given twice daily. Although the overall response rate is low for fluoxymesterone used in this clinical situation,190 there are some patients who have substantial antitumor responses lasting for months or even years.
Toxicities associated with fluoxymesterone are those that would be expected with an androgen: hirsutism, male-pattern baldness, voice lowering (hoarseness), acne, enhanced libido, and erythrocytosis. Fluoxymesterone can also cause elevated liver function test results in some patients and, rarely, has been associated with hepatic neoplasms.
Pharmacology
Fluoxymesterone is a chlorinated synthetic analog of testosterone with potent androgenic and anabolic activity in humans. Limited pharmacologic information is available on this agent. Colburn,191 using a radioimmunoassay, studied two patients after a single oral administration of a 50-mg dose. Peak serum concentrations were achieved between 1 and 3 hours after administration, with the average peak concentrations being 335 ng/mL. By 5 hours after drug administration, serum levels had declined to approximately 50% of the peak concentration. Urinary excretion of a 10-mg dose can be detected for 24 hours, and at least 6-hydroxy, 4-ene, 3-β, and 11-hydroxy metabolites of fluoxymesterone have been detected.192
Estrogens: Diethylstilbestrol and Estradiol
Diethylstilbestrol (DES) had been the primary hormonal therapy for postmenopausal metastatic breast cancer. Randomized comparative trials demonstrated it had a similar response rate to that of tamoxifen.193,194 However, based on these trials, DES use was supplanted by tamoxifen, primarily because DES has more toxicity. DES is occasionally used in metastatic breast cancer patients who have hormonally sensitive cancers that have failed to respond to multiple other hormonal therapies. The usual dose in this situation is 15 mg per day, either as a single dose or as divided doses. DES was also used as androgen ablation therapy in men with metastatic prostate cancer.195 Doses of approximately 3 mg per day result in testosterone levels that are seen in an anorchid state.
DES toxicities include nausea and vomiting, breast tenderness, and a darkening of the nipple–areolar complex. DES increases the risk of thromboembolic phenomenon, which may result in life-threatening complications. Although DES is not clinically available in the United States, similar antitumor effects and toxicities are seen with estradiol, with a target dose of 10 mg by mouth three times a day. The pharmacology of E2 has been extensively described elsewhere.196
Medroxyprogesterone and Megestrol
Medroxyprogesterone and megestrol are 17-OH-progesterone derivatives differing in a double bond between C6 and C7 positions in megestrol. Historically, megestrol was used as a hormonal agent for patients with advanced breast cancer, usually at a total daily dose of 160 mg. Additionally, it is still used for the treatment of hormonally responsive metastatic endometrial cancer, at a dose of 320 mg per day. In addition, doses of 160 mg per day are occasionally used as a hormonal therapy for prostate cancer.197 Megestrol has also been extensively evaluated for the treatment of anorexia/cachexia related to cancer or AIDS.198–201 Various dosages ranging from 160 to 1,600 mg per day have been used. A prospective study has demonstrated a dose–response relationship with doses up to 800 mg per day.202 Low dosages of megestrol (20 to 40 mg per day) have been shown to be an effective means of reducing hot flashes in women with breast cancer and in men who have undergone androgen ablation therapy.5 Although megestrol had historically been commonly administered four times per day, the long terminal half-life supports once-per-day dosing.
Megestrol is a relatively well-tolerated medication, with its most prominent side effects being appetite stimulation and resultant weight gain. Although these may be beneficial effects in patients with anorexia/cachexia, they can be important problems in patients with breast or endometrial cancers. Another side effect of megestrol acetate is the marked suppression of adrenal steroid production by suppression of the pituitary–adrenal axis.203 Although this appears to be asymptomatic in the majority of patients, reports suggest that this adrenal suppression can cause clinical problems in some patients.204This drug has been abruptly stopped for decades without the recognition of untoward sequelae in patients, and it seems reasonable to continue this practice. Nonetheless, if Addisonian signs or symptoms develop after drug discontinuation, corticosteroids should be administered. Furthermore, if patients who receive megestrol have a significant infection, experience trauma, or undergo surgery, then corticosteroid coverage should be administered. There appears to be a slightly increased incidence of thromboembolic phenomena in patients receiving megestrol alone.202 This risk appears to be higher if megestrol is administered with concomitant cytotoxic therapy.205 There are conflicting reports regarding megestrol-causing edema.206 If it does, the edema is generally minimal and easily handled with a mild diuretic. Megestrol may cause impotence in some men.207 The incidence of this is controversial, although it is generally agreed that this is a reversible situation. Megestrol can cause menstrual irregularities, the most prominent of which is withdrawal menstrual bleeding within a few weeks of drug discontinuation.5 Although nausea and vomiting have sometimes been attributed as a toxicity of this drug, there are data to demonstrate that this drug has antiemetic properties.200,201,205 In terms of magnitude, megestrol appears to decrease both nausea and vomiting in advanced-stage cancer patients by approximately two thirds.
Medroxyprogesterone has many of the same properties, clinical uses, and toxicities as megestrol acetate. It has never been commonly used in the United States for the treatment of breast cancer but has been used more in Europe. Medroxyprogesterone is available in 2.5- and 10-mg tablets and in injectable formulations of 100 and 400 mg/L. Dosing for the treatment of metastatic breast or prostate cancer has commonly been 400 mg per week or more and 1,000 mg per week or more for metastatic endometrial cancer. Injectable or daily oral doses have been used for controlling hot flashes.
Pharmacology
The exact mechanism of antitumor effect of medroxyprogesterone and megestrol is unclear. These drugs have been reported to suppress adrenal steroid synthesis,208 suppress ER levels,209 alter tumor hormone metabolism,210 enhance steroid metabolism,211 and directly kill tumor cells.212 In addition, progestins may influence some growth factors,213 suppress plasma estrone sulfate formation, and, at high concentrations, inhibit P-glycoprotein.
The oral bioavailability of these progestational agents is unknown, although absorption appears to be poor for medroxyprogesterone relative to megestrol.
The terminal half-life for megestrol is approximately 14 hours,214,215 with a tmax of 2 to 5 hours after oral ingestion.216 The AUC for a single megestrol dose of 160 mg is between 2.5- and 8-fold higher than that for single-dose medroxyprogesterone at 1,000 mg with a radioactive dose of megestrol; 50% to 78% is found in the urine after oral administration, and 8% to 30% is found in the feces.
Metabolism and excretion of medroxyprogesterone have been incompletely characterized. In humans, 20% to 50% of a [3H]medroxyprogesterone dose is excreted in the urine and 5% to 10% in the stool after intravenous administration.217–219 Metabolism of medroxyprogesterone occurs via hydroxylation, reduction, demethylation, and combinations of these reactions.220 The major urinary metabolite is a glucuronide. Less than 3% of the dose is excreted as unconjugated medroxyprogesterone in humans. Clearance of medroxyprogesterone has been reported to range between 27 and 70 L per hour.219 The initial volume of distribution is between 4 and 8 L in humans. The mean terminal half-life is 60 hours. The tmax for medroxyprogesterone occurs 2 to 5 hours after oral administration. Medroxyprogesterone appears to be concentrated in the small intestine, the colon, and in adipose tissue in human autopsy studies.221 Drug interactions of medroxyprogesterone have been reported with aminoglutethimide, which decreases plasma medroxyprogesterone levels.222 Medroxyprogesterone may reduce the concentration of the N-desmethyltamoxifen metabolite concentration. Progestational agents also may increase plasma warfarin levels.223 These reports are consistent with CYP3A being the site of interaction.
OTHER HORMONAL THERAPIES
Octreotide
Octreotide is a somatostatin analog that is administered for the treatment of carcinoid syndrome and other hormonal excess syndromes associated with some pancreatic islet cell cancers and acromegaly. Response rates (measured in terms of a reduction in diarrhea and flushing) are high and can last for several months to years. Occasionally, antitumor responses temporarily related to octreotide are seen with these tumors. Octreotide may be useful to alleviate 5-fluorouracil–associated diarrhea.224–226
Octreotide can be administered intravenously or subcutaneously. Initial doses of 50 μg are given two to three times on the first day. The dose is titrated upward, with a usual daily dose of 300 to 450 μg per day for most patients. A depot preparation is available, allowing doses to be administered at monthly intervals. Octreotide is generally well tolerated overall. It appears to cause more toxicity in acromegalic patients, with such problems as bradycardia, diarrhea, hypoglycemia, hyperglycemia, hypothyroidism, and cholelithiasis.
Pharmacology
Octreotide is an 8-amino acid synthetic analog of the 14-amino acid peptide somatostatin.227 Octreotide has a similar high affinity for somatostatin receptors, as does its parent compound, with a concentration that inhibits the receptor by 50% in the subnanomolar range. Octreotide inhibits insulin, glucagon, pancreatic polypeptide, gastric inhibitory polypeptide, and gastrin secretion. It has a much longer duration of action than the parent compound because of its greater resistance to enzymatic degradation. Its absorption after subcutaneous administration is rapid, and bioavailability is 100% after subcutaneous injection. Peak concentrations of 4 μg/L after a 100-μg dose occur within 20 to 30 minutes of subcutaneous injection and are 20% to 40% of the corresponding intravenous injection. Both peak concentration and AUC for octreotide increase linearly with dose. The total body clearance in healthy volunteers is 9.6 L per hour. Hepatic metabolism of octreotide accounts for 30% to 40% of the drug’s disposition, and 11% to 20% is excreted unchanged in the urine. The volume of distribution ranges between 18 and 30 L, and the terminal half-life is reported to be between 72 and 98 minutes. Sixty-five percent of the drug is protein bound primarily to the lipoprotein fraction.227,228 Because of the short half-life, classic octreotide is administered subcutaneously two or three times per day.229 A slow-release form of octreotide, designed for once-per-month administration, controls the symptoms of carcinoid syndrome at least as well as three-times-per-day octreotide.230
REFERENCES
1. Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst 2005;97:1652–1662.
2. Fisher B, Dignam J, Wolmark N, et al. Tamoxifen in treatment of intraductal breast cancer: National Surgical Adjuvant Breast and Bowel Project B-24 randomised controlled trial. Lancet 1999;353:1993–2000.
3. Colleoni M, Gelber S, Goldhirsch A, et al. Tamoxifen after adjuvant chemotherapy for premenopausal women with lymph node-positive breast cancer: International Breast Cancer Study Group Trial 13-93. J Clin Oncol 2006;24:1332–1341.
4. Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805–816.
5. Loprinzi CL, Michalak JC, Quella SK, et al. Megestrol acetate for the prevention of hot flashes. N Engl J Med 1994;331:347–352.
6. Loprinzi CL, Kugler JW, Sloan JA, et al. Venlafaxine in management of hot flashes in survivors of breast cancer: a randomised controlled trial. Lancet 2000;356:2059–2063.
7. Archer DF, Dupont CM, Constantine GD, et al. Desvenlafaxine for the treatment of vasomotor symptoms associated with menopause: a double-blind, randomized, placebo-controlled trial of efficacy and safety. Am J Obstet Gynecol 2009;200:238.e1–238.e10.
8. Barton DL, LaVasseur BI, Sloan JA, et al. Phase III, placebo-controlled trial of three doses of citalopram for the treatment of hot flashes: NCCTG trial N05C9. J Clin Oncol 2010;28:3278–3283.
9. Freeman EW, Guthrie KA, Caan B, et al. Efficacy of escitalopram for hot flashes in healthy menopausal women: a randomized controlled trial. JAMA 2011;305:267–274.
10. Stearns V, Beebe KL, Iyengar M, et al. Paroxetine controlled release in the treatment of menopausal hot flashes: a randomized controlled trial. JAMA 2003;289:2827–2834.
11. Pandya KJ, Morrow GR, Roscoe JA, et al. Gabapentin for hot flashes in 420 women with breast cancer: a randomised double-blind placebo-controlled trial. Lancet 2005;366:818–824.
12. Loprinzi CL, Qin R, Balcueva EP, et al. Phase III, randomized, double-blind, placebo-controlled evaluation of pregabalin for alleviating hot flashes, N07C1. J Clin Oncol 2010;28:641–647.
13. Madlensky L, Natarajan L, Tchu S, et al. Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin Pharmacol Ther 2011;89:718–725.
14. Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet 1998;351:1451–1467.
15. Wickerham DL, Fisher B, Wolmark N, et al. Association of tamoxifen and uterine sarcoma. J Clin Oncol 2002;20:2758–2760.
16. Dewar JA, Horobin JM, Preece PE, et al. Long term effects of tamoxifen on blood lipid values in breast cancer. BMJ 1992;305:225–226.
17. Love RR, Mazess RB, Barden HS, et al. Effects of tamoxifen on bone mineral density in postmenopausal women with breast cancer. N Engl J Med 1992;326:852–856.
18. Powles TJ, Hickish T, Kanis JA, et al. Effect of tamoxifen on bone mineral density measured by dual-energy x-ray absorptiometry in healthy premenopausal and postmenopausal women. J Clin Oncol 1996;14:78–84.
19. Buzdar A, Howell A, Cuzick J, et al. Comprehensive side-effect profile of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: long-term safety analysis of the ATAC trial. Lancet Oncol 2006;7:633–643.
20. Gorin MB, Day R, Costantino JP, et al. Long-term tamoxifen citrate use and potential ocular toxicity. Am J Ophthalmol 1998;125:493–501.
21. Tonetti DA, Jordan VC. Possible mechanisms in the emergence of tamoxifen-resistant breast cancer. Anticancer Drugs 1995;6:498–507.
22. Lim YC, Desta Z, Flockhart DA, et al. Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother Pharmacol 2005;55:471–478.
23. Benz CC, Scott GK, Sarup JC, et al. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res Treat 1993;24:85–95.
24. Borg A, Baldetorp B, Ferno M, et al. ERBB2 amplification is associated with tamoxifen resistance in steroid-receptor positive breast cancer. Cancer Lett 1994;81:137–144.
25. Houston SJ, Plunkett TA, Barnes DM, et al. Overexpression of c-erbB2 is an independent marker of resistance to endocrine therapy in advanced breast cancer. Br J Cancer 1999;79:1220–1226.
26. Lipton A, Ali SM, Leitzel K, et al. Serum HER-2/neu and response to the aromatase inhibitor letrozole versus tamoxifen. J Clin Oncol 2003;21:1967–1972.
27. Bunone G, Briand PA, Miksicek RJ, et al. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. Embo J 1996;15:2174–2183.
28. Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995;270:1491–1494.
29. Pietras RJ, Arboleda J, Reese DM, et al. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene 1995;10:2435–2446.
30. Osborne CK, Bardou V, Hopp TA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 2003;95:353–361.
31. Encarnacion CA, Ciocca DR, McGuire WL, et al. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat 1993;26:237–246.
32. Watts CK, Handel ML, King RJ, et al. Oestrogen receptor gene structure and function in breast cancer. J Steroid Biochem Mol Biol 1992;41:529–536.
33. Zhang QX, Borg A, Wolf DM, et al. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 1997;57:1244–1249.
34. Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45:1439–1445.
35. Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013;45:1446–1451.
36. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, et al. D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 2013;73:6856–6864.
37. Fendl KC, Zimniski SJ. Role of tamoxifen in the induction of hormone-independent rat mammary tumors. Cancer Res 1992;52:235–237.
38. Williams GM. Tamoxifen experimental carcinogenicity studies: implications for human effects. Proc Soc Exp Biol Med 1995;208:141–143.
39. Williams GM, Iatropoulos MJ, Djordjevic MV, et al. The triphenylethylene drug tamoxifen is a strong liver carcinogen in the rat. Carcinogenesis 1993;14:315–317.
40. Rutqvist LE, Johansson H, Signomklao T, et al. Adjuvant tamoxifen therapy for early stage breast cancer and second primary malignancies. Stockholm Breast Cancer Study Group. J Natl Cancer Inst 1995;87:645–651.
41. Mani C, Kupfer D. Cytochrome P-450-mediated activation and irreversible binding of the antiestrogen tamoxifen to proteins in rat and human liver: possible involvement of flavin-containing monooxygenases in tamoxifen activation. Cancer Res 1991;51:6052–6058.
42. Mani C, Pearce R, Parkinson A, et al. Involvement of cytochrome P4503A in catalysis of tamoxifen activation and covalent binding to rat and human liver microsomes. Carcinogenesis 1994;15:2715–2720.
43. Styles JA, Davies A, Lim CK, et al. Genotoxicity of tamoxifen, tamoxifen epoxide and toremifene in human lymphoblastoid cells containing human cytochrome P450s. Carcinogenesis 1994;15:5–9.
44. Han XL, Liehr JG. Induction of covalent DNA adducts in rodents by tamoxifen. Cancer Res 1992;52:1360–1363.
45. Mani C, Hodgson E, Kupfer D.Metabolism of the antimammary cancer antiestrogenic agent tamoxifen. II. Flavin-containing monooxygenase-mediated N-oxidation. Drug Metab Dispos 1993;21:657–661.
46. Mani C, Gelboin HV, Park SS, et al. Metabolism of the antimammary cancer antiestrogenic agent tamoxifen. I. Cytochrome P-450-catalyzed N-demethylation and 4-hydroxylation. Drug Metab Dispos 1993;21:645–656.
47. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol2010;11:55–65.
48. Dowsett M, Cuzick J, Wale C, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol2010;28:1829–1834.
49. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–2826.
50. Johnson MD, Zuo H, Lee KH, et al. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res Treat 2004;85:151–159.
51. Lim YC, Li L, Desta Z, et al. Endoxifen, a secondary metabolite of tamoxifen, and 4-OH-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J Pharmacol Exp Ther 2006;318:503–512.
52. Wu X, Hawse JR, Subramaniam M, et al. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res 2009;69:1722–1727.
53. Jin Y, Desta Z, Stearns V, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst 2005;97:30–39.
54. Desta Z, Ward BA, Soukhova NV, et al. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther 2004;310:1062–1075.
55. Stearns V, Johnson MD, Rae JM, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst 2003;95:1758–1764.
56. Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005;23:9312–9318.
57. Schroth W, Antoniadou L, Fritz P, et al. Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. J Clin Oncol 2007;25:5187–5193.
58. Schroth W, Goetz MP, Hamann U, et al. Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA 2009;302:1429–1436.
59. Rae JM, Drury S, Hayes DF, et al. CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J Natl Cancer Inst 2012;104:452–460.
60. Regan MM, Leyland-Jones B, Bouzyk M, et al. CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the breast international group 1-98 trial. J Natl Cancer Inst 2012;104:441–451.
61. Goetz MP, Suman VJ, Hoskin TL, et al. CYP2D6 metabolism and patient outcome in the Austrian Breast and Colorectal Cancer Study Group trial (ABCSG) 8. Clin Cancer Res 2013;19:500–507.
62. Province MA, Goetz MP, Brauch H, et al. CYP2D6 Genotype and adjuvant tamoxifen: meta-analysis of heterogeneous study populations. Clin Pharmacol Ther 2014;95:216–227.
63. Borges S, Desta Z, Li L, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther 2006;80:61–74.
64. Dezentje VO, van Blijderveen NJ, Gelderblom H, et al. Effect of concomitant CYP2D6 inhibitor use and tamoxifen adherence on breast cancer recurrence in early-stage breast cancer. J Clin Oncol 2010;28:2423–2429.
65. Kelly CM, Juurlink DN, Gomes T, et al. Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: a population based cohort study. BMJ 2010;340:c693.
66. Binkhorst L, van Gelder T, Loos WJ, et al. Effects of CYP induction by rifampicin on tamoxifen exposure. Clin Pharmacol Ther 2012;92:62–67.
67. Irvin WJ Jr., Walko CM, Weck KE, et al. Genotype-guided tamoxifen dosing increases active metabolite exposure in women with reduced CYP2D6 metabolism: a multicenter study. J Clin Oncol 2011;29:3232–3239.
68. Kiyotani K, Mushiroda T, Imamura CK, et al. Dose-adjustment study of tamoxifen based on CYP2D6 genotypes in Japanese breast cancer patients. Breast Cancer Res Treat 2012;131:137–145.
69. Goetz MP, Suman VA, Reid JR, et al. A first-in-human phase I study of the tamoxifen (TAM) metabolite, Z-endoxifen hydrochloride (Z-Endx) in women with aromatase inhibitor (AI) refractory metastatic breast cancer (MBC) (NCT01327781). Cancer Res 2013;73(24 Suppl): Abstract nr PD3-4.
70. Lien EA, Anker G, Lonning PE, et al. Decreased serum concentrations of tamoxifen and its metabolites induced by aminoglutethimide. Cancer Res 1990;50:5851–5857.
71. Adam HK, Patterson JS, Kemp JV. Studies on the metabolism and pharmacokinetics of tamoxifen in normal volunteers. Cancer Treat Rep 1980;64:761–764.
72. Patterson JS, Settatree RS, Adam HK, et al. Serum concentrations of tamoxifen and major metabolite during long-term nolvadex therapy, correlated with clinical response. Eur J Cancer Suppl 1980;1:89–92.
73. Camaggi CM, Strocchi E, Canova N, et al. Medroxyprogesterone acetate (MAP) and tamoxifen (TMX) plasma levels after simultaneous treatment with ’low’ TMX and ’high’ MAP doses. Cancer Chemother Pharmacol 1985;14:229–231.
74. Lien EA, Solheim E, Lea OA, et al. Distribution of 4-hydroxy-N-desmethyltamoxifen and other tamoxifen metabolites in human biological fluids during tamoxifen treatment. Cancer Res 1989;49:2175–2183.
75. Lien EA, Solheim E, Ueland PM. Distribution of tamoxifen and its metabolites in rat and human tissues during steady-state treatment. Cancer Res 1991;51:4837–4844.
76. Daniel P, Gaskell SJ, Bishop H, et al. Determination of tamoxifen and biologically active metabolites in human breast tumours and plasma. Eur J Cancer Clin Oncol 1981;17:1183–1189.
77. Robinson SP, Langan-Fahey SM, Johnson DA, et al. Metabolites, pharmacodynamics, and pharmacokinetics of tamoxifen in rats and mice compared to the breast cancer patient. Drug Metab Dispos 1991;19:36–43.
78. DeGregorio MW, Wiebe VJ, Venook AP, et al. Elevated plasma tamoxifen levels in a patient with liver obstruction. Cancer Chemother Pharmacol 1989;23:194–195.
79. Lodwick R, McConkey B, Brown AM. Life threatening interaction between tamoxifen and warfarin. Br Med J (Clin Res Ed) 1987;295:1141.
80. Ritchie LD, Grant SM. Tamoxifen-warfarin interaction: the Aberdeen hospitals drug file. BMJ 1989;298:1253.
81. Tenni P, Lalich DL, Byrne MJ. Life threatening interaction between tamoxifen and warfarin. BMJ 1989;298:93.
82. Rabinowicz AL, Hinton DR, Dyck P, et al. High-dose tamoxifen in treatment of brain tumors: interaction with antiepileptic drugs. Epilepsia 1995;36:513–515.
83. Desai PB, Nallani SC, Sane RS, et al. Induction of cytochrome P450 3A4 in primary human hepatocytes and activation of the human pregnane X receptor by tamoxifen and 4-hydroxytamoxifen. Drug Metab Dispos 2002;30:608–612.
84. Dowsett M, Cuzick J, Howell A, et al. Pharmacokinetics of anastrozole and tamoxifen alone, and in combination, during adjuvant endocrine therapy for early breast cancer in postmenopausal women: a sub-protocol of the ‘Arimidex and tamoxifen alone or in combination’ (ATAC) trial. Br J Cancer2001;85:317–324.
85. Pagani O, Gelber S, Price K, et al. Toremifene and tamoxifen are equally effective for early-stage breast cancer: first results of International Breast Cancer Study Group Trials 12-93 and 14-93. Ann Oncol 2004;15:1749–1759.
86. Hayes DF, Van Zyl JA, Hacking A, et al. Randomized comparison of tamoxifen and two separate doses of toremifene in postmenopausal patients with metastatic breast cancer. J Clin Oncol 1995;13:2556–2566.
87. Stenbygaard LE, Herrstedt J, Thomsen JF, et al. Toremifene and tamoxifen in advanced breast cancer—a double-blind cross-over trial. Breast Cancer Res Treat 1993;25:57–63.
88. Vogel CL, Shemano I, Schoenfelder J, et al. Multicenter phase II efficacy trial of toremifene in tamoxifen-refractory patients with advanced breast cancer. J Clin Oncol 1993;11:345–350.
89. Kangas L. Review of the pharmacological properties of toremifene. J Steroid Biochem 1990;36:191–195.
90. Berthou F, Dreano Y, Belloc C, et al. Involvement of cytochrome P450 3A enzyme family in the major metabolic pathways of toremifene in human liver microsomes. Biochem Pharmacol 1994;47:1883–1895.
91. Simberg NH, Murai JT, Siiteri PK. In vitro and in vivo binding of toremifene and its metabolites in rat uterus. J Steroid Biochem 1990;36:197–202.
92. Kohler PC, Hamm JT, Wiebe VJ, et al. Phase I study of the tolerance and pharmacokinetics of toremifene in patients with cancer. Breast Cancer Res Treat 1990;16 Suppl:S19–S26.
93. Tominaga T, Abe O, Izuo M. A phase I study of toremifene. Breast Cancer Res Treat 1990;16 (Suppl):27.
94. Wiebe VJ, Benz CC, Shemano I, et al. Pharmacokinetics of toremifene and its metabolites in patients with advanced breast cancer. Cancer Chemother Pharmacol 1990;25:247–251.
95. Anttila M, Laakso S, Nylanden P, et al. Pharmacokinetics of the novel antiestrogenic agent toremifene in subjects with altered liver and kidney function. Clin Pharmacol Ther 1995;57:628–635.
96. Hard GC, Iatropoulos MJ, Jordan K, et al. Major difference in the hepatocarcinogenicity and DNA adduct forming ability between toremifene and tamoxifen in female Crl:CD(BR) rats. Cancer Res 1993;53:4534–4541.
97. Montandon F, Williams GM. Comparison of DNA reactivity of the polyphenylethylene hormonal agents diethylstilbestrol, tamoxifen and toremifene in rat and hamster liver. Arch Toxicol 1994;68:272–275.
98. Vogel VG, Costantino JP, Wickerham DL, et al. Update of the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 Trial: Preventing Breast Cancer. Cancer Prev Res (Phila) 2010;3:696–706.
99. Delmas PD, Balena R, Confravreux E, et al. Bisphosphonate risedronate prevents bone loss in women with artificial menopause due to chemotherapy of breast cancer: a double-blind, placebo-controlled study. J Clin Oncol 1997;15:955–962.
100. Draper MW, Flowers DE, Huster WJ, et al. A controlled trial of raloxifene (LY139481) HCl: impact on bone turnover and serum lipid profile in healthy postmenopausal women. J Bone Miner Res 1996;11:835–842.
101. Anzano MA, Peer CW, Smith JM, et al. Chemoprevention of mammary carcinogenesis in the rat: combined use of raloxifene and 9-cis-retinoic acid. J Natl Cancer Inst 1996;88:123–125.
102. Gottardis MM, Jordan VC. Antitumor actions of keoxifene and tamoxifen in the N-nitrosomethylurea-induced rat mammary carcinoma model. Cancer Res 1987;47:4020–4024.
103. Black LJ, Jones CD, Falcone JF. Antagonism of estrogen action with a new benzothiophene derived antiestrogen. Life Sci 1983;32:1031–1036.
104. Allerheiligen S, Geiser J, Knadler M. Raloxifen (RAL) pharmacokinetics and the associated endocrine effects in premenopausal women treated during the follicular, ovulatory, and luteal phases of the menstrual cycle. Pharmaceut Res 1996;13:S430.
105. Forgue ST, Rudy AC, Knadler MP. Raloxifene pharmacokinetics in healthy postmenopausal women. Pharmaceut Res 1996;13:S430.
106. Ni L, Allerheiligen S, Basson R. Pharacokinetics of raloxifene in men and postmenopausal women volunteers. Pharmaceut Res 1996;13:S430.
107. Kemp DC, Fan PW, Stevens JC. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug Metab Dispos 2002;30:694–700.
108. Coopman P, Garcia M, Brunner N, et al. Anti-proliferative and anti-estrogenic effects of ICI 164,384 and ICI 182,780 in 4-OH-tamoxifen-resistant human breast-cancer cells. Int J Cancer 1994;56:295–300.
109. Howell A, DeFriend DJ, Robertson JF, et al. Pharmacokinetics, pharmacological and anti-tumour effects of the specific anti-oestrogen ICI 182780 in women with advanced breast cancer. Br J Cancer 1996;74:300–308.
110. Howell A, Osborne CK, Morris C, et al. ICI 182,780 (Faslodex): development of a novel, “pure” antiestrogen. Cancer 2000;89:817–825.
111. Robertson JF, Odling-Smee W, Holcombe C, et al. Pharmacokinetics of a single dose of fulvestrant prolonged-release intramuscular injection in postmenopausal women awaiting surgery for primary breast cancer. Clin Ther 2003;25:1440–1452.
112. Piccart M, Parker LM, Pritchard KI. Oestrogen receptor downregulation: an opportunity for extending the window of endocrine therapy in advanced breast cancer. Ann Oncol 2003;14:1017–1025.
113. Wakeling AE, Bowler J. Steroidal pure antioestrogens. J Endocrinol 1987;112:R7–R10.
114. Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res 1991;51:3867–3873.
115. Howell A, Robertson JF, Quaresma Albano J, et al. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol 2002;20:3396–3403.
116. Osborne CK, Pippen J, Jones SE, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol 2002;20:3386–3395.
117. Howell A, Robertson JF, Abram P, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol2004;22:1605–1613.
118. Leo AD, Jerusalem G, Petruzelka L, et al. Final overall survival: fulvestrant 500 mg vs 250 mg in the randomized CONFIRM trial. J Natl Cancer Inst 2014;106:djt337.
119. McCormack P, Sapunar F. Pharmacokinetic profile of the fulvestrant loading dose regimen in postmenopausal women with hormone receptor-positive advanced breast cancer. Clin Breast Cancer 2008;8:347–351.
120. Schteingart DE, Cash R, Conn JW. Amino-glutethimide and metastatic adrenal cancer. Maintained reversal (six months) of Cushing’s syndrome. JAMA 1996;198:1007–1010.
121. Ma CX, Adjei AA, Salavaggione OE, et al. Human aromatase: gene resequencing and functional genomics. Cancer Res 2005;65:11071–11082.
122. Colomer R, Monzo M, Tusquets I, et al. A single-nucleotide polymorphism in the aromatase gene is associated with the efficacy of the aromatase inhibitor letrozole in advanced breast carcinoma. Clin Cancer Res 2008;14:811–816.
123. Wang L, Ellsworth KA, Moon I, et al. Functional genetic polymorphisms in the aromatase gene CYP19 vary the response of breast cancer patients to neoadjuvant therapy with aromatase inhibitors. Cancer Res 2010;70:319–328.
124. Goss PE, Ingle JN, Martino S, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med 2003;349:1793–1802.
125. Mouridsen H, Gershanovich M, Sun Y, et al. Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol2003;21:2101–2109.
126. Howell A, Cuzick J, Baum M, et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 2005;365:60–62.
127. Thurlimann B, Keshaviah A, Coates AS, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med 2005;353:2747–2757.
128. Jakesz R, Jonat W, Gnant M, et al. Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years’ adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet 2005;366:455–462.
129. Cuzick J, Sestak I, Forbes JF, et al. Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II): an international, double-blind, randomised placebo-controlled trial. Lancet 2014;383:1041–1048.
130. Baum M, Budzar AU, Cuzick J, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet 2002;359:2131–2139.
131. Amir E, Seruga B, Nira S, et al. Toxicity of adjuvant endocrine therapy in postmenopausal breast cancer patients: a systematic review and meta-analysis. J Natl Cancer Inst 2011;103:1299–1309.
132. Plourde PV, Dyroff M, Dukes M. Arimidex: a potent and selective fourth-generation aromatase inhibitor. Breast Cancer Res Treat 1994;30:103–111.
133. Bisagni G, Cocconi G, Scaglione F, et al. Letrozole, a new oral non-steroidal aromastase inhibitor in treating postmenopausal patients with advanced breast cancer. A pilot study. Ann Oncol 1996;7:99–102.
134. Buzdar AU. Pharmacology and pharmacokinetics of the newer generation aromatase inhibitors. Clin Cancer Res 2003;9:468S–472S.
135. Dewar JA, Nabholtz JM, Bonneterre J, et al. The effect of anastrozole (Arimidex) on serum lipids: data from a randomized comparison of anastrozole (AN) versus tamoxifen (TAM) in postmenopausal (PM) women with advanced breast cancer (ABC). Breast Cancer Res Treat 2000;64:51.
136. Elisaf MS, Bairaktari ET, Nicolaides C, et al. Effect of letrozole on the lipid profile in postmenopausal women with breast cancer. Eur J Cancer 2001;37:1510–1513.
137. Bhatnagar AS, Hausler A, Schieweck K. Inhibition of aromatase in vitro and in vivo by aromatase inhibitors. J Enzyme Inhib 1990;4:179–186.
138. Bhatnagar AS, Hausler A, Schieweck K, et al. Highly selective inhibition of estrogen biosynthesis by CGS 20267, a new non-steroidal aromatase inhibitor. J Steroid Biochem Mol Biol 1990;37:1021–1027.
139. Demers LM. Effects of Fadrozole (CGS 16949A) and Letrozole (CGS 20267) on the inhibition of aromatase activity in breast cancer patients. Breast Cancer Res Treat 1994;30:95–102.
140. Lipton A, Demers LM, Harvey HA, et al. Letrozole (CGS 20267). A phase I study of a new potent oral aromatase inhibitor of breast cancer. Cancer 1995;75:2132–2138.
141. Iveson TJ, Smith IE, Ahern J, et al. Phase I study of the oral nonsteroidal aromatase inhibitor CGS 20267 in postmenopausal patients with advanced breast cancer. Cancer Res 1993;53:266–270.
142. Trunet PF, Muller PH, Bhatnagar A. Phase I study in healthy male volunteers with the non-steroidal aromatase inhibitor GCS 20267. Eur J Cancer 1990;26:173.
143. Dukes M, Edwards PN, Large M, et al. The preclinical pharmacology of “Arimidex” (anastrozole; ZD1033)—a potent, selective aromatase inhibitor. J Steroid Biochem Mol Biol 1996;58:439–445.
144. Yates RA, Dowsett M, Fisher GV, et al. Arimidex (ZD1033): a selective, potent inhibitor of aromatase in postmenopausal female volunteers. Br J Cancer 1996;73:543–548.
145. Lonning PE, Geisler J, Dowsett M. Pharmacological and clinical profile of anastrozole. Breast Cancer Res Treat 1998;49:S53–S57.
146. Geisler J, King N, Dowsett M, et al. Influence of anastrozole (Arimidex), a selective, non-steroidal aromatase inhibitor, on in vivo aromatisation and plasma oestrogen levels in postmenopausal women with breast cancer. Br J Cancer 1996;74:1286–1291.
147. Ingle JN, Buzdar AU, Schaid DJ, et al. Variation in anastrozole metabolism and pharmacodynamics in women with early breast cancer. Cancer Res 2010;70:3278–3286.
148. Kaufmann M, Bajetta E, Dirix LY, et al. Exemestane is superior to megestrol acetate after tamoxifen failure in postmenopausal women with advanced breast cancer: results of a phase III randomized double-blind trial. The Exemestane Study Group. J Clin Oncol2000;18:1399–1411.
149. Goss PE, Ingle JN, Pritchard KI, et al. Exemestane versus anastrozole in postmenopausal women with early breast cancer: NCIC CTG MA.27—a randomized controlled phase III trial. J Clin Oncol 2013;31:1398–1404.
150. Goss PE, Ingle JN, Ales-Martinez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med 2011;364:2381–2391.
151. Goss PE, Grynpas M, Qi S, et al. The effects of exemestane on bone and lipids in the ovariectomized rat. Breast Cancer Res Treat 2001;69:224.
152. Evans TR, Di Salle E, Ornati G, et al. Phase I and endocrine study of exemestane (FCE 24304), a new aromatase inhibitor, in postmenopausal women. Cancer Res 1992;52:5933–5939.
153. Bajetta E, Zilembo N, Noberasco C, et al. The minimal effective exemestane dose for endocrine activity in advanced breast cancer. Eur J Cancer 1997;33:587–591.
154. Michaud LB, Buzdar AU. Risks and benefits of aromatase inhibitors in postmenopausal breast cancer. Drug Saf 1999;21:297–309.
155. Coombes RC, Hall E, Gibson LJ, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med 2004;350:1081–1092.
156. Demers LM, Lipton A, Harvey HA, et al. The efficacy of CGS 20267 in suppressing estrogen biosynthesis in patients with advanced stage breast cancer. J Steroid Biochem Mol Biol 1993;44:687–691.
157. Spinelli R, Jannuzzo MG, Poggesi I, et al. Pharmacokinetics (PK) of Aromasin (Exemestane, EXE) after single and repeated doses in healthy postmenopausal volunteers (HPV). Eur J Cancer 1999;35:S295.
158. Buzdar A, Howell A. Advances in aromatase inhibition: clinical efficacy and tolerability in the treatment of breast cancer. Clin Cancer Res 2001;7:2620–2635.
159. Johannessen DC, Engan T, Di Salle E, et al. Endocrine and clinical effects of exemestane (PNU 155971), a novel steroidal aromatase inhibitor, in postmenopausal breast cancer patients: a phase I study. Clin Cancer Res 1997;3:1101–1108.
160. Jones S, Vogel C, Arkhipov A, et al. Multicenter, phase II trial of exemestane as third-line hormonal therapy of postmenopausal women with metastatic breast cancer. Aromasin Study Group. J Clin Oncol 1999;17:3418–3425.
161. Ahmann FR, Citrin DL, deHaan HA, et al. Zoladex: a sustained-release, monthly luteinizing hormone-releasing hormone analogue for the treatment of advanced prostate cancer. J Clin Oncol 1987;5:912–917.
162. Corbin A. From contraception to cancer: a review of the therapeutic applications of LHRH analogues as antitumor agents. Yale J Biol Med 1982;55:27–47.
163. Kaufmann M, Jonat W, Blamey R, et al. Survival analyses from the ZEBRA study. Goserelin (Zoladex) versus CMF in premenopausal women with node-positive breast cancer. Eur J Cancer 2003;39:1711–1717.
164. Harvey HA, Lipton A, Max DT, et al. Medical castration produced by the GnRH analogue leuprolide to treat metastatic breast cancer. J Clin Oncol 1985;3:1068–1072.
165. Abrahamsson PA. Potential benefits of intermittent androgen suppression therapy in the treatment of prostate cancer: a systematic review of the literature. Eur Urol 2010;57:49–59.
166. Hussain M, Tangen CM, Berry DL, et al. Intermittent versus continuous androgen deprivation in prostate cancer. N Engl J Med 2013;368:1314–1325.
167. Brogden RN, Faulds D. Goserelin. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in prostate cancer. Drugs Aging 1995;6:324–343.
168. Vogelzang NJ, Chodak GW, Soloway MS, et al. Goserelin versus orchiectomy in the treatment of advanced prostate cancer: final results of a randomized trial. Zoladex Prostate Study Group. Urology 1995;46:220–226.
169. Plosker GL, Brogden RN. Leuprorelin. A review of its pharmacology and therapeutic use in prostatic cancer, endometriosis and other sex hormone-related disorders. Drugs 1994;48:930–967.
170. Nillius SJ. The Therapeutic Uses of Gonadotropin-Releasing Hormone and Its Analogues. London: Butterworth; 1981.
171. Klijn JG, DeJong FH, Blankenstein MA. Anti-tumor and endocrine effects of chronic LHRH agonist treatment (buserelin) with or without tamoxifen in premenopausal metastatic breast cancer. Breast Cancer Res Treat 1984;4:209.
172. Clayton RN, Bailey LC, Cottam J, et al. A radioimmunoassay for GnRH agonist analogue in serum of patients with prostate cancer treated with D-Ser (tBu)6 AZA Gly10 GnRH. Clin Endocrinol (Oxf) 1985;22:453–462.
173. Chrisp P, Goa KL. Goserelin. A review of its pharmacodynamic and pharmacokinetic properties, and clinical use in sex hormone-related conditions. Drugs 1991;41:254–288.
174. Samant MP, Hong DJ, Croston G, et al. Novel gonadotropin-releasing hormone antagonists with substitutions at position 5. Biopolymers 2005;80:386–391.
175. Van Poppel H, Tombal B, de la Rosette JJ, et al. Degarelix: a novel gonadotropin-releasing hormone (GnRH) receptor blocker—results from a 1-yr, multicentre, randomised, phase 2 dosage-finding study in the treatment of prostate cancer. Eur Urol 2008;54:805–813.
176. Klotz L, Boccon-Gibod L, Shore ND, et al. The efficacy and safety of degarelix: a 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU Int 2008;102:1531–1538.
177. Steinberg M. Degarelix: a gonadotropin-releasing hormone antagonist for the management of prostate cancer. Clin Ther 2009;31:2312–2331.
178. Brogden RN, Clissold SP. Flutamide. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in advanced prostatic cancer. Drugs 1989;38:185–203.
179. Wysowski DK, Freiman JP, Tourtelot JB, et al. Fatal and nonfatal hepatotoxicity associated with flutamide. Ann Intern Med 1993;118:860–864.
180. Brogden RN, Chrisp P. Flutamide. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in advanced prostatic cancer. Drugs Aging 1991;1:104–115.
181. Radwanski E, Perentesis G, Symchowicz S, et al. Single and multiple dose pharmacokinetic evaluation of flutamide in normal geriatric volunteers. J Clin Pharmacol 1989;29:554–558.
182. Schellhammer PF, Sharifi R, Block NL, et al. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate carcinoma. Analysis of time to progression. CASODEX Combination Study Group. Cancer1996;78:2164–2169.
183. Furr BJ. Casodex (ICI 176,334)—a new, pure, peripherally-selective anti-androgen: preclinical studies. Horm Res 1989;32:69.
184. Furr BJ. Casodex: preclinical studies. Eur Urol 1990;18:2.
185. Kennealey GT, Furr BJ. Use of the nonsteroidal anti-androgen Casodex in advanced prostatic carcinoma. Urol Clin North Am 1991;18:99–110.
186. Attard G, Reid AH, A’Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol 2009;27:3742–3748.
187. Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787–790.
188. Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187–1197.
189. Zhu H, Garcia JA. Targeting the adrenal gland in castration-resistant prostate cancer: a case for orteronel, a selective CYP-17 17,20-lyase inhibitor. Curr Oncol Rep 2013;15:105–112.
190. Kennedy BJ. Hormonal therapies in breast cancer. Semin Oncol 1974;1:119–130.
191. Colburn WA. Radioimmunoassay for fluoxymesterone (Halotestin). Steroids 1975;25:43–52.
192. Kammerer RC, Merdink JL, Jagels M, et al. Testing for fluoxymesterone (Halotestin) administration to man: identification of urinary metabolites by gas chromatography-mass spectrometry. J Steroid Biochem 1990;36:659–666.
193. Ingle JN, Ahmann DL, Green SJ, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med 1981;304:16–21.
194. Stewart HJ, Forrest AP, Gunn JM, et al. The tamoxifen trial - a double-blind comparison with stilboestrol in postmenopausal women with advanced breast cancer. Eur J Cancer 1980;Suppl 1:83–88.
195. Byar DP. Proceedings: The Veterans Administration Cooperative Urological Research Group’s studies of cancer of the prostate. Cancer 1973;32:1126–1130.
196. Loose-Mitchell DS, Stancel GM. Estrogens and Progestins. 10 ed. New York: McGraw-Hill; 2001.
197. Bonomi P, Pessis D, Bunting N, et al. Megestrol acetate used as primary hormonal therapy in stage D prostatic cancer. Semin Oncol 1985;12:36–39.
198. Bruera E, Macmillan K, Kuehn N, et al. A controlled trial of megestrol acetate on appetite, caloric intake, nutritional status, and other symptoms in patients with advanced cancer. Cancer 1990;66:1279–1282.
199. Feliu J, Gonzalez-Baron M, Berrocal A, et al. Usefulness of megestrol acetate in cancer cachexia and anorexia. A placebo-controlled study. Am J Clin Oncol 1992;15:436–440.
200. Loprinzi CL, Ellison NM, Schaid DJ, et al. Controlled trial of megestrol acetate for the treatment of cancer anorexia and cachexia. J Natl Cancer Inst 1990;82:1127–1132.
201. Tchekmedyian NS, Hickman M, Siau J, et al. Megestrol acetate in cancer anorexia and weight loss. Cancer 1992;69:1268–1274.
202. Loprinzi CL, Michalak JC, Schaid DJ, et al. Phase III evaluation of four doses of megestrol acetate as therapy for patients with cancer anorexia and/or cachexia. J Clin Oncol 1993;11:762–767.
203. Loprinzi CL, Jensen MD, Jiang NS, et al. Effect of megestrol acetate on the human pituitary-adrenal axis. Mayo Clin Proc 1992;67:1160–1162.
204. Leinung MC, Liporace R, Miller CH. Induction of adrenal suppression by megestrol acetate in patients with AIDS. Ann Intern Med 1995;122:843–845.
205. Rowland KM Jr., Loprinzi CL, Shaw EG, et al. Randomized double-blind placebo-controlled trial of cisplatin and etoposide plus megestrol acetate/placebo in extensive-stage small-cell lung cancer: a North Central Cancer Treatment Group study. J Clin Oncol 1996;14:135–141.
206. Loprinzi CL, Johnson PA, Jensen M. Megestrol acetate for anorexia and cachexia. Oncology 1992;49:46–49.
207. Von Roenn JH, Armstrong D, Kotler DP, et al. Megestrol acetate in patients with AIDS-related cachexia. Ann Intern Med 1994;121:393–399.
208. Alexieva-Figusch J, Blankenstein MA, Hop WC, et al. Treatment of metastatic breast cancer patients with different dosages of megestrol acetate; dose relations, metabolic and endocrine effects. Eur J Cancer Clin Oncol 1984;20:33–40.
209. Tseng L, Gurpide E. Effects of progestins on estradiol receptor levels in human endometrium. J Clin Endocrinol Metab 1975;41:402–404.
210. Gurpide E, Tseng L, Gusberg SB. Estrogen metabolism in normal and neoplastic endometrium. Am J Obstet Gynecol 1977;129:809–816.
211. Gordon GG, Altman K, Southren AL, et al. Human hepatic testosterone A-ring reductase activity: effect of medroxyprogesterone acetate. J Clin Endocrinol Metab 1971;32:457–461.
212. Allegra JC, Kiefer SM. Mechanisms of action of progestational agents. Semin Oncol 1985;12:3–5.
213. Ewing TM, Murphy LJ, Ng ML, et al. Regulation of epidermal growth factor receptor by progestins and glucocorticoids in human breast cancer cell lines. Int J Cancer 1989;44:744–752.
214. Adlercreutz H, Eriksen PB, Christensen MS. Plasma concentration of megestrol acetate and medroxyprogesterone acetate after single oral administration to healthy subjects. J Pharm Biomed Anal 1983;1:153.
215. Martin F, Adlercreutz H, eds. Aspects of Megestrol Acetate and Medroxyprogesterone Acetate Metabolism. New York: Raven Press; 1977.
216. Gaver RC, Pittman KA, Reilly CM, et al. Bioequivalence evaluation of new megestrol acetate formulations in humans. Semin Oncol 1985;12:17–19.
217. Fotherby K, Kamyab S, Littleton P. Metabolism of synthetic progestational compounds in humans. J Reprod Fertil 1968;5:51–61.
218. Fukushima DK, Ievin J, Liang JS, et al. Isolation and partial synthesis of a new metabolite of medroxyrogesterone acetate. Steroids 1979;34:57–72.
219. Utaaker E, Lundgren S, Kvinnsland S, et al. Pharmacokinetics and metabolism of medroxyprogesterone acetate in patients with advanced breast cancer. J Steroid Biochem 1988;31:437–441.
220. Sturm G, Haberlein H, Bauer T, et al. Mass spectrometric and high-performance liquid chromatographic studies of medroxyprogesterone acetate metabolites in human plasma. J Chromatogr 1991;562:351–362.
221. Pannuti F, Camaggi CM, Strocchi E, eds. Medroxyprogesterone Acetate Pharmacokinetics. New York: Raven Press; 1984.
222. Lundgren S, Lonning PE, Aakvaag A, et al. Influence of aminoglutethimide on the metabolism of medroxyprogesterone acetate and megestrol acetate in postmenopausal patients with advanced breast cancer. Cancer Chemother Pharmacol 1990;27:101–105.
223. Lundgren S, Kvinnsland S, Utaaker E, et al. Effect of oral high-dose progestins on the disposition of antipyrine, digitoxin, and warfarin in patients with advanced breast cancer. Cancer Chemother Pharmacol 1986;18:270–275.
224. Cascinu S, Fedeli A, Fedeli SL, et al. Control of chemotherapy-induced diarrhoea with octreotide in patients receiving 5-fluorouracil. Eur J Cancer 1992;28:482–483.
225. Cascinu S, Fedeli A, Fedeli SL, et al. Octreotide versus loperamide in the treatment of fluorouracil-induced diarrhea: a randomized trial. J Clin Oncol 1993;11:148–151.
226. Gebbia V, Carreca I, Testa A, et al. Subcutaneous octreotide versus oral loperamide in the treatment of diarrhea following chemotherapy. Anticancer Drugs 1993;4:443–445.
227. Harris AG. Somatostatin and somatostatin analogues: pharmacokinetics and pharmacodynamic effects. Gut 1994;35:S1–S4.
228. Chanson P, Timsit J, Harris AG. Clinical pharmacokinetics of octreotide. Therapeutic applications in patients with pituitary tumours. Clin Pharmacokinet 1993;25:375–391.
229. Marbach P, Briner U, Lemaire M. From somatostatin to Sandostatin: pharmacodynamics and pharmacokinetics. Digestion 1993;54:9–13.
230. Rubin J, Ajani J, Schirmer W, et al. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J Clin Oncol 1999;17:600–606.