Ephraim Paul Hochberg
INTRODUCTION
Metabolic emergencies represent rare events in the care of cancer patients. In contrast to the measured pace of most of oncology, these scenarios demand prompt identification and intervention. This chapter reviews tumor lysis syndrome, hyponatremia, and hypercalcemia.
TUMOR LYSIS SYNDROME
![]() DEFINITION
DEFINITION
Tumor lysis syndrome is a collection of metabolic derangements secondary to the release of tumor cell contents into the extracellular space (Table 20-1). The rapid development of hyperphosphatemia, hyperuricemia, and hyperkalemia can cause devastating renal and cardiac complications. Hypocalcemia occurs secondarily as a consequence of hyperphosphatemia.
TABLE 20-1 FEATURES OF TUMOR LYSIS SYNDROME

Tumor lysis syndrome occurs most frequently in rapidly growing hematologic malignancies such as acute leukemias and Burkitt’s lymphoma. In these diseases, it can occur either spontaneously or more commonly 6–72 h following initiation of antitumor therapy. There are case reports of even more rapid onset with the use of targeted therapies. The syndrome is most frequently associated with cytoxic chemotherapy but can occur after embolization, radiation, or glucocorticoids. The Cairo-Bishop definitions and grading system for laboratory and clinical tumor lysis syndrome are not widely used in clinical practice (Table 20-2) (1). The National Cancer Institute Common Toxicity Criteria 4.0 system grade TLS by its presence (grade 3), life threatening consequences (grade 4), or death (grade 5).
TABLE 20-2 CAIRO AND BISHOP DEFINITION AND GRADING CLASSIFICATION OF TUMOR LYSIS SYNDROME

![]() INCIDENCE AND RISK FACTORS
INCIDENCE AND RISK FACTORS
There are both tumor-related risk factors for tumor lysis syndrome, as well as patient-specific risks. Tumor-specific risks include a large burden of disease, a high-proliferative rate, or a highly treatment-sensitive tumor. Clinically significant tumor lysis syndrome is estimated to occur in up to 30% of patients with Burkitt’s lymphoma/B-ALL and 15% of patients with acute myeloid leukemia, although laboratory evidence of tumor lysis syndrome can be present in a larger proportion of patients. There is a significant but lower risk in diffuse large B-cell lymphoma, and rarer reports in chronic lymphocytic leukemia, and multiple myeloma. The use of the monoclonal antibody rituximab has been associated with the development of tumor lysis syndrome in patients with increased numbers of circulating tumor cells (≥25,000) or a large tumor burden. Tumor lysis syndrome is rare in solid tumors, with breast cancer and small cell lung cancer comprising the most common reports. There are case reports of tumor lysis syndrome with a number of targeted therapies (Table 20-3).
TABLE 20-3 RISK FACTORS FOR TUMOR LYSIS SYNDROME

Patient comorbidities increase the risk of tumor lysis syndrome. Decreased urinary flow or dehydration, chronic renal insufficiency or frank renal failure, acidic urine, or preexisting hyperuricemia are all risk factors for tumor lysis syndrome development.
![]() MECHANISMS
MECHANISMS
Purine Catabolism
The major contributing element in the pathophysiology of tumor lysis syndrome is hyperuricemia caused by the release of nucleic acids (purines), which are catabolized to uric acid, leading to elevated plasma urate levels (Figure 20-1). Elevated uric acid levels overwhelm the excretory capacity of the renal tubules particularly in the presence of an acidic pH and low urine flow. Crystalline obstruction of the renal tubules, uric acid nephropathy, renal failure, and uremia may result. Renal failure may also occur due to volume depletion and changes in autoregulation within the kidney.
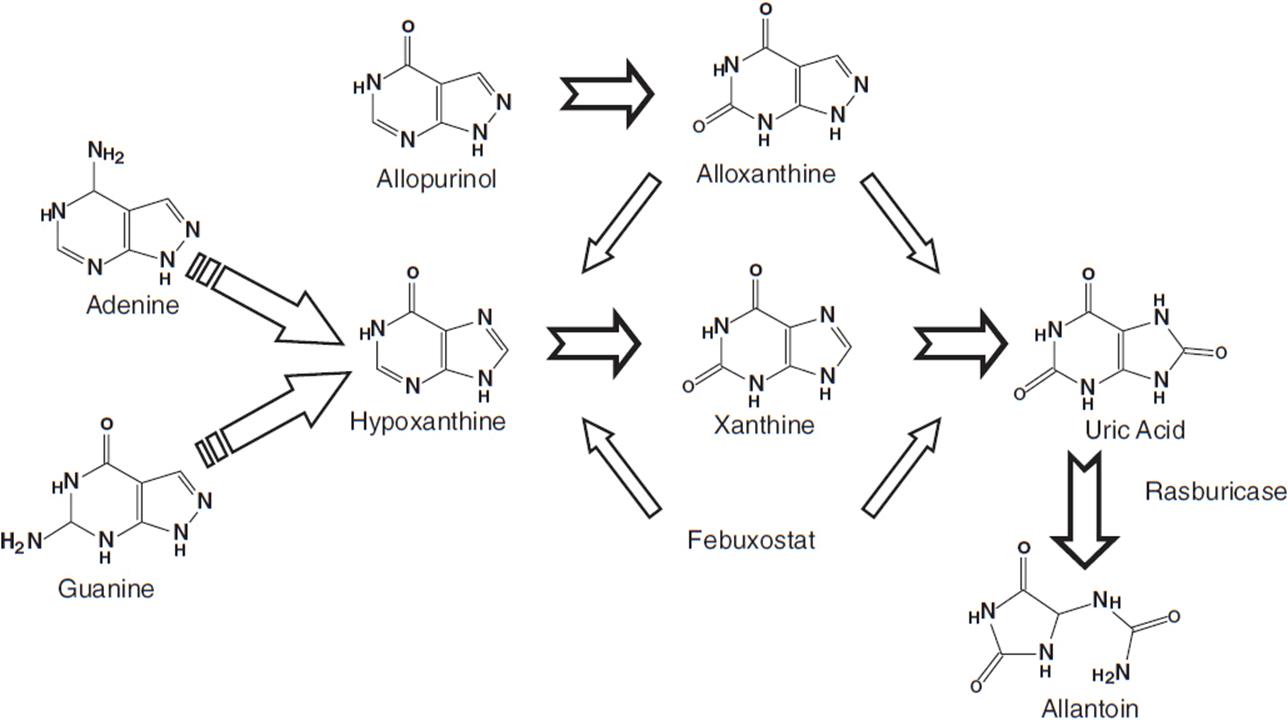
FIGURE 20-1 Uric acid formation via purine catabolism and the mechanism of action of allopurinol, rasburicase, and febuxostat.
Intracellular Ion Release
Other intracellular ions are released, including potassium and phosphorus. Hyperkalemia-induced cardiac arrhythmias exacerbated by renal failure are the most life-threatening complication of tumor lysis syndrome.
Intracellular phosphorous, which may be overproduced in malignant cells, is also released, renally excreted, and may precipitate as calcium phosphate in the renal tubules, leading to hypocalcemia, metastatic calcification, intrarenal calcification, nephrocalcinosis, nephrolithiasis, and obstructive uropathy.
Hypocalcemia is generally due to precipitation of calcium phosphate in the soft tissues and kidneys during periods of hyperphosphatemia and inappropriately low levels of 1, 25-dihydroxyvitamin D3. This can be clinically associated with tetany and cardiac arrhythmias.
![]() CLINICAL PRESENTATION
CLINICAL PRESENTATION
The clinical presentation of tumor lysis syndrome is characterized by the symptoms of the individual electrolyte imbalances and resultant renal failure (Table 20-4).
TABLE 20-4 CLINICAL SIGNS AND SYMPTOMS OF TUMOR LYSIS SYNDROME

![]() DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of tumor lysis syndrome is limited. Injuries to large structures such as ischemic limb or rhabdomyolysis can replicate the varied clinical and laboratory presentations of tumor lysis syndrome but should be clinically apparent. The other laboratory abnormalities may be present to some extent in patients with renal insufficiency from any cause (Table 20-5).
TABLE 20-5 DIFFERENTIAL DIAGNOSIS OF TUMOR LYSIS SYNDROME

![]() THERAPEUTIC AGENTS, MONITORING
THERAPEUTIC AGENTS, MONITORING
See Tables 20-3 and 20-6 for an overview of the following discussion. Patient- and tumor-specific factors are combined in a stratification schema that divides patients into high, intermediate, and low-risk cohorts. High-risk patients include all patients with Burkitt’s lymphoma, lymphoblastic lymphoma, or B-cell ALL as well as patients with ALL with a WBC ≥100,000, AML with WBC ≥50,000, or monoblastic subtype. Intermediate-risk patients include all diffuse large B-cell lymphoma, ALL with WBC 50–100,000, AML with WBC 10–50,000, and CLL with WBC ≥10,000 treated with fludarabine as well as all other rapidly proliferating malignancies with an expected rapid response to therapy. Low-risk patients include all indolent lymphomas as well as ALL with a WBC ≤50,000, AML and CLL with WBC ≤10,000 (2).
TABLE 20-6 OVERVIEW OF TUMOR LYSIS PROPHYLAXIS AND TREATMENT

In patients at high risk of developing tumor lysis syndrome, the decision to delay therapy must be weighed against the risks of tumor lysis syndrome, but in any case prophylactic measures should be instituted. Drugs that contribute to electrolyte abnormalities are nephrotoxic, or block uric acid, or potassium excretion should be stopped.
Hydration and Diuresis
The mainstay of prevention is aggressive hydration and diuresis, with individuals receiving 2–4 times usual maintenance fluids or 3–6 l/m2/day. These fluids should not initially include potassium, calcium, or phosphate. If the patient is not hypovolemic but is oligo or anuric, diuretics such as furosemide (or mannitol, particularly if furosemide fails) may be used to maintain an appropriate urine output with the following as goals: urine specific gravity should be <1.010 and urine output >150–250 ml/h. Furosemide diuresis may also decrease serum potassium levels. Dosing of diuretics is patient dependent and may be intermittent or continuous.
Traditionally, recommendations have also suggested alkalinization of urine with sodium bicarbonate. However, this is not recommended in the era of recombinant urate oxidase due to the increased risk of urinary xanthine and calcium phosphate deposition, fluid overload, and metabolic alkalosis (3).
Allopurinol
Allopurinol, a xanthine analog that is converted to alloxanthine and acts as a competitive inhibitor of xanthine oxidase, reduces the production of uric acid and the incidence of related obstructive uropathy (Figure 20-1). It does not reduce levels of uric acid already present and leads to lower urate levels over days. Allopurinol may also result in xanthine nephropathy and calculi due to accumulation of xanthine and hypoxanthine, which are renally excreted.
Usual dosing of allopurinol is 2 mg/m2/day or 600–800 mg/day orally, 12 h to 3 days prior to initiation of chemotherapy (or 200–400 mg/m2/day IV, 24–48 h before initiation of chemotherapy; maximum dose 600 mg/day if unable to take orally). Allopurinol should be renally dosed in appropriate patients. The intravenous and oral forms have equivalent efficacy.
Allopurinol also reduces the degradation of other purines including 6-mercaptopurine (6-MP) and azothioprine, and therefore, these drugs should be dose-reduced when used with allopurinol.
Rasburicase
Rasburicase (Elitek) is a recombinant urate oxidase that converts uric acid to allantoin, a more soluble compound, which is then renally excreted (Figure 20-1). Usual dosing is 0.20 mg/kg IV over 30 min for 1–5 days and does not require renal dosing. Side effects include anaphylaxis or hypersensitivity reactions (bronchospasm, hypoxemia) in 5% of those who receive this drug. In addition, patients may form antibodies to this drug, the significance of which is currently unknown. Rasburicase should not be used in patients with G-6PD deficiency as hemolytic anemia and methemoglobinemia may occur. In the majority of patients, a single dose is sufficient to control hyperuricemia.
A randomized trial comparing rasburicase, allopurinol, and the combination of the two agents in adult and pediatric patients at high risk for tumor lysis syndrome demonstrated more rapid control of uric acid in a higher percentage of patients in the group treated with rasburicase alone.
Febuxostat
Febuxostat is a nonpurine selective inhibitor of xanthine oxidase used for prevention of gout. A phase 3 trial for tumor lysis syndrome prevention is currently ongoing.
Monitoring
Monitoring for tumor lysis involves twice daily serum electrolytes (more frequently if the patient is at high risk or clinically unstable), and at least daily urinary pH and spot uric acid to creatinine ratio (value should be >1.0), evaluation of volume status (including daily weights, blood pressure, and examination looking for edema, pleural effusions, and ascites), and serum WBC count, LDH, ionized or corrected calcium, and calcium phosphorus product.
ECG changes associated with hyperkalemia include widening of the QRS complex (late manifestations include a sine wave appearance, ventricular arrhythmias, or asystole) and peaked T waves, while hypocalcemia is associated with a long QT secondary to a prolonged ST segment.
![]() TREATMENT
TREATMENT
Hyperkalemia
Hyperkalemia treatment can be divided into treatment for moderate or asymptomatic hyperkalemia and those for severe or symptomatic hyperkalemia (Table 20-6).
For asymptomatic or moderate hyperkalemia, oral sodium polystyrene sulphonate (Kayexalate), an exchange resin, is the primary treatment (15 g orally 1–4 times daily as a slurry in water or 70% sorbitol or as a retention enema, 30–50 g rectally every 6 h as a warm emulsion in 100 ml aqueous vehicle (sorbitol or D20W), retained for 30–60 min in adults).
For severe cases, Kayexalate in addition to insulin and dextrose, sodium bicarbonate (1 mEq/kg, repeated as needed based on blood gas values), loop diuretics, inhaled beta-agonists, and, in extreme examples, calcium gluconate or chloride is appropriate. For calcium gluconate, the dosage is 2–4 mg/kg of a 10% solution, repeated at 10-min intervals as necessary. Calcium chloride is reserved for emergency situations. Hemodialyis or hemofiltration should also be considered.
Hyperuricemia And Renal Failure
Hyperuricemia should be treated with rasburicase. Hemodialysis or hemofiltration may be necessary.
Renal failure requires aggressive fluid, electrolyte, and uric acid management along with close monitoring for indications requiring hemodialysis or hemofiltration. Hemodialysis indications may include fluid overload, metabolic acidosis, electrolyte disturbances that are recalcitrant to the treatments outlined above, uremia, and hypertension.
Hyperphosphatemia
Hyperphosphatemia is treated with oral aluminum hydroxide (adult dosing: 500–1800 mg, 3–6 times daily between meals) or aluminum carbonate (2 capsules or tablets or 12 ml suspension 3–4 times daily with meals). Insulin and dextrose as administered in hyperkalemia can also be given. Severe or symptomatic cases should receive hemodialysis or hemofiltration.
Hypocalcemia
Moderate or asymptomatic hypocalcemia usually does not require treatment, and the risks of treatment may outweigh the benefits. In severe cases, calcium gluconate 500 mg to 2 g (10% solution) IV in adults at a rate not to exceed 1.5 ml/min can be used but may lead to calcium phosphate precipitation resulting obstructive uropathy or metastatic calcification. In an emergency situation, calcium chloride should be used for hyperkalemia or hypocalcemia at 2–4 mg/kg of a 10% solution, repeated at 10-min intervals as necessary.
HYPONATREMIA
![]() INCIDENCE AND RISK FACTORS
INCIDENCE AND RISK FACTORS
Hyponatremia is a common electrolyte abnormality, occurring in many elderly and/or hospitalized patients. In cancer patients, hyponatremia may be due to primary or metastatic disease, cancer-related medical interventions or complications, usually via the syndrome of inappropriate antidiuretic hormone (SIADH), as well as other causes. This section will focus primarily on the diagnosis and treatment of SIADH-related hyponatremia in the setting of malignancy (4).
Malignancy-related hyponatremia is most commonly seen in lung carcinomas, particularly the small cell variant. However, head and neck cancers, leukemia, and mediastinal cancers have also been implicated, along with limited reports in a number of other cancers. Hyponatremia can also be therapy related and has been reported after stem cell transplantation.
![]() MECHANISMS
MECHANISMS
Hyponatremia can be characterized by varying levels of osmolality (Table 20-7). Hypotonic hyponatremia, the most common form, is caused by abnormal retention of water with varying amounts of fluid intake (Table 20-8). Retention of water occurs because the ability of the kidneys to excrete fluid has been impaired or overwhelmed. Edema of the central nervous system is the most concerning consequence of hyponatremia.
TABLE 20-7 TYPES OF HYPONATREMIA WITH MECHANISM AND REPRESENTATIVE CAUSES

TABLE 20-8 CAUSES AND TREATMENT OF HYPOTONIC HYPONATREMIA BY EXTRACELLULAR FLUID VOLUME, TOTAL BODY WATER, AND SODIUM STORES

Other uncommon causes of hypotonic hyponatremia, not included in Table 20-7, include excessive water intake either orally (as in polydipsia) or via absorption of sodium-free fluids (irrigant solutions, enemas) or reset osmostat syndrome; the latter is a subset of SIADH.
Mechanism Of Siadh
SIADH occurs because of paraneoplastic or ectopic production of antidiuretic hormone (ADH) or arginine vasopressin (AVP). AVP binds to the V2 vasopressin receptor (V2R), a G protein-coupled receptor, in the collecting duct and the ascending limb of the loop of Henle. This leads to increases in intracellular cyclic AMP (c-AMP) that in turn leads to increased water permeability, inappropriate water retention, and, therefore, hyponatremia and hypo-osmolality. Patients with SIADH may exhibit inappropriate thirst (5).
Normally, AVP or V2 vasopressin is produced in the supraoptic and paraventricular nuclei of the hypothalamus and transported down the neurohypophysis, or posterior lobe of the pituitary gland, where it is released into the blood stream when cardiovascular baro- and osmoreceptors signal that blood pressure or plasma volume are too low or plasma osmolality is too high. AVP then acts on the kidney as described above, to retain water and concentrate urine, and may have a direct, pressor effect on arteries and arterioles.
However, the etiology of malignancy-associated hyponatremia may not be due only to ectopic production of AVP (Tables 20-7, 20-8, and 20-9). Consideration should be given to other causes of hyponatremia or SIADH, as this will affect treatment.
TABLE 20-9 DIFFERENTIAL DIAGNOSIS OF MALIGNANCY-ASSOCIATED HYPONATREMIA

![]() DIAGNOSIS
DIAGNOSIS
Variable definitions for hyponatremia are used and make comparison of cases difficult. In this chapter, hyponatremia is defined as serum sodium concentration below 135 mmol/l (this is consistent with our institutional range, 135–145 mmol/l, of serum sodium). Diagnostic testing is based upon the hypothesized cause of hyponatremia. For SIADH, the diagnosis should also be based on the criteria in Tables 20-10 and 20-11. AVP level need not be checked. The clinical signs of hyponatremia are listed in Table 20-12.
TABLE 20-10 CLINICAL FEATURES AND RESPONSE TO CLINICAL CHALLENGES BY TYPE OF HYPONATREMIA

TABLE 20-11 DIAGNOSTIC FACTORS SUGGESTING SYNDROME OF INAPPROPRIATE ANTIDIURETIC HORMONE (SIADH)

TABLE 20-12 CLINICAL SIGNS AND SYMPTOMS OF HYPONATREMIA

![]() PROPHYLAXIS AND MONITORING
PROPHYLAXIS AND MONITORING
There is no prophylaxis for hyponatremia other than to anticipate electrolyte abnormalities, actively replete electrolyte losses, and avoid drugs that induce hyponatremia.
![]() TREATMENT
TREATMENT
General Comments
Treatment of the underlying disease is the most important therapy. For SIADH tumor response is correlated with resolution of hyponatremia. If other factors, such as drugs, diet, or free water consumption, are worsening the condition, appropriate adjustments should be made.
Hypokalemia is often present with hyponatremia, and correction of hypokalemia will increase serum sodium either via osmotic shifts of sodium and free water or urinary losses. Administration of potassium therefore must be taken into account (see the formula below). Hyperkalemia, without obvious cause, in the presence of hyponatremia warrants consideration of adrenal insufficiency.
Treatment of hypotonic hyponatremia is dependent upon the cause, severity, chronicity, and presence or absence of symptoms. In general, correction of hyponatremia, regardless of etiology, should not exceed 0.5 mM/h or more than 10–15 mEq/l in 24 h unless severe symptoms are present (seizures, coma). In severe cases with acute onset of hyponatremia, rates of correction of 1–3 mM/h have been recommended for short periods. Central pontine myelinolysis can complicate rapid correction (see below).
![]() TREATMENT OF SIADH
TREATMENT OF SIADH
The initial approach to mild, asymptomatic hyponatremia is fluid restriction to 800 ml/day or a negative water balance.
Symptomatic or moderate to severe hyponatremia will likely require hypertonic saline administration. Isotonic saline should not be used in SIADH. The formula below can assist in determining the expected rise in serum sodium to a given quantity of fluid repletion.

See Table 20-13. Total body water (TBW) is 0.6 in men and children, and 0.5 for women. These values are slightly lower for older individuals. Based on the goal rate of change, the rate of fluid administration can be calculated.
TABLE 20-13 FLUID CHOICE FOR CORRECTION OF HYPONATREMIA

Another method to determine the amount of saline necessary is to calculate the patient’s sodium deficit and then the amount and rate of fluid administration needed to correct this deficit using the formula below:

The desired serum sodium is generally 120–125 mmol/l as correction to strictly normal levels of serum sodium via hypertonic saline is generally not necessary or desirable.
Diuresis (with furosemide) may be used to maintain appropriate volume status and to inhibit sodium chloride reabsorption in the kidney and thereby aid in sodium correction, but should be used cautiously to avoid overly rapid correction. Serum sodium should be monitored every 2 h initially and adjustments made accordingly.
Salt supplementation (high-salt diet, salt tablets), loop diuretics, and water restriction are employed in the long-term management for SIADH once severe hyponatremia has been corrected.
Drug Therapies
The vasopressin receptor antagonists produce water diuresis without effecting sodium excretion. Tolvaptan (po) and conivaptan (IV) are currently available in the United States. These agents are limited by the risk of over-rapid correction of serum sodium but can be highly effective in treating hyponatremia. Demeclocycline, a tetracycline antibiotic that can block AVP activation of renal c-AMP, induces a state of nephrogenic diabetes insipidus. It can be given as 600–1200 mg/day in 2–3 divided doses if the above methods fail or cannot be tolerated. Demeclocycline dose should be adjusted for renal and hepatic dysfunction. Photosensitivity, nausea, and nephrotoxicity may occur and it may take days to see a response. Lithium, urea, and fludrocortisone are other medications that have been used in refractory cases.
![]() TREATMENT OF OTHER CAUSES OF HYPONATREMIA
TREATMENT OF OTHER CAUSES OF HYPONATREMIA
Initial techniques for mild, hypervolemic hyponatremia include both fluid and salt restriction. Further treatment must be based on the underlying etiology of the hyponatremia.
Patients who have hypovolemic hyponatremia with sodium loss may benefit from larger volumes of 0.9% sodium chloride initially, especially if blood pressure is low. However, these patients must be monitored closely as they approach euvolemia, as they tend to excrete water more rapidly than sodium as the impetus for ADH release declines, leading to more rapid sodium correction.
Reset osmostat, which is a special case of SIADH, presents as a mild, stable hyponatremia that generally does not require correction and is best treated by treating the underlying condition.
Treatment of hypertonic hyponatremia should be directed at the underlying cause.
![]() TREATMENT COMPLICATIONS
TREATMENT COMPLICATIONS
A feared complication of overrapid sodium repletion is cerebral myelinolysis. This condition has been described most in the pons where it presents as a rapidly progressive weakness of limbs combined with dysarthria, dysphagia, seizures, and death occurring 2–6 days after correction, with CT and MRI evidence developing 6–10 days after clinical symptoms.
HYPERCALCEMIA
![]() INCIDENCE AND RISK FACTORS
INCIDENCE AND RISK FACTORS
Normal values for serum calcium levels are population, laboratory, and laboratory machinery specific. However, one author defined moderate hypercalcemia as total serum calcium (adjusted for albumin) greater than 12 mg/dl and severe as greater than 14 mg/dl. At our institution, normal values for serum calcium are 8.5–10.5 mg/dl and for ionized calcium are 1.14–1.30 mmol/l.
Hypercalcemia may occur in as many as 30% of cancer cases. Among solid tumors, breast cancer, non-small cell lung cancer, squamous cell cancers, head and neck cancer, and renal cancers are highest risk; while among hematologic malignancies, multiple myeloma and lymphoma patients are at the highest risk. Hypercalcemia is uncommonly seen in other solid malignancies, such as colon, prostate, and small cell lung cancer.
The risk for hypercalcemia is determined by the histology and location of the cancer, the duration of illness, and the site of metastases. Risk of death is reported to be higher in individuals with malignancy-associated hypercalcemia.
![]() MECHANISMS
MECHANISMS
Normally PTH and vitamin D act on bone, kidney, and gut to maintain serum calcium levels. Hypercalcemia occurs when either intestinal absorption of calcium increases, bone resorption increases, renal reabsorption increases, or release of tumor-related humoral factors leads to increased serum calcium levels that exceed renal excretion. The two most common causes of malignancy-associated hypercalcemia are local, osteolytic activity of tumor cells and humoral hypercalcemia of malignancy. These two etiologies represent a continuum of malignancy-related pathology (6).
Osteolytic Hypercalcemia
Osteolytic hypercalcemia comprises 20% of cases and is commonly due to breast cancer, myeloma, and lymphoma with substantial bone involvement. Release of osteoclast activating factors including lymphotoxin, IL-1 alpha, TGF alpha and beta, TNF alpha, and IL-6 have been identified, among others. In osteolytic hypercalcemia, phosphate is usually normal, while 1, 25(OH)2 vitamin D3, intestinal reabsorption of calcium and PTH are low, and renal clearance of calcium is increased.
Humoral Hypercalcemia of Malignancy
The other 80% of hypercalcemia cases are due to humoral hypercalcemia via parathyroid hormone related protein (PTHrP) and are usually associated with squamous cell cancers; renal, breast, ovarian, or endometrial cancers; and HTLV-associated ATLL (Acute T cell leukemia/lymphoma). Release of PTHrP appears to contribute to osteolysis locally and systemically by increasing osteoclast activity and therefore bone resorption. Due to close homology with PTH, PTHrP mimics the effect of PTH on renal and skeletal calcium homeostasis.
Humoral hypercalcemia should be considered when PTH levels are low, when metastases are absent, and when metabolic alkalosis with low chloride and high bicarbonate concentrations are present. In addition, 1, 25(OH)2vitamin D3 and phosphorus levels are generally low, while urinary c-AMP concentrations and renal calcium clearance are high.
Other Mechanisms of Hypercalcemia
There are further rare causes of malignancy-associated hypercalcemia that represent a small fraction of cases. These are included in Table 20-14.
TABLE 20-14 RARE CAUSES OF HYPERCALCEMIA IN MALIGNANCY

![]() CLINICAL PRESENTATION
CLINICAL PRESENTATION
The symptoms of hypercalcemia can be remarkably protean with effects on numerous organ systems (Table 20-15). The presence or absence of symptoms is dependent upon individual patient factors such as severity, chronicity, preexisting mental status, age, concomitant sedatives, or narcotics.
TABLE 20-15 CLINICAL SIGNS AND SYMPTOMS OF HYPERCALCEMIA

![]() DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
The most frequent causes of hypercalcemia are cancer, primary hyperparathyroidism, and vitamin D intoxication (Table 20-16).
TABLE 20-16 DIFFERENTIAL DIAGNOSIS FOR HYPERCALCEMIA

![]() DIAGNOSIS
DIAGNOSIS
Ideally ionized calcium would be drawn, given variations in calcium level by albumin level and the presence of calcium-binding immunoglobulins. If total serum calcium levels are used, correct for albumin level via the following formula:

In addition to calcium, serum electrolytes, phosphorus, BUN and creatinine, PTH, and PTHrP (as well as SPEP, UPEP, free serum kappa and lambda light chains, and Bence Jones proteins if myeloma is suspected) are often measured in malignancy-associated hypercalcemia. The presence of bone metastases on radionuclide imaging may also help confirm the diagnosis. Distinguishing between rare causes of malignancy-related hypercalcemia and other causes usually requires further testing, such as vitamin D levels, thyroid function tests, fasting calcium to creatinine urine ratio, chest x-ray, and serum ACE levels.
![]() PROPHYLAXIS AND MONITORING
PROPHYLAXIS AND MONITORING
There are no clear guidelines for the frequency of calcium monitoring. However, it should be based, at least in part, on the expected length of efficacy of any treatment rendered. For instance, for bisphosphonates, the treatment effect generally lasts a month or less. Measurement of serum or ionized calcium, urea and other electrolytes, albumin, volume status including urine output, and mental status are typical.
![]() TREATMENT
TREATMENT
The primary treatment of malignancy-associated hypercalcemia is directed toward the underlying cancer. However, Table 20-17 summarizes treatments frequently used as temporizing measures.
TABLE 20-17 TREATMENT FOR MALIGNANCY-RELATED HYPERCALCEMIA

First-Line Treatments
Hydration and Diuresis Mild hypercalcemia can be treated with saline hydration, and, when appropriate, diuresis. Most patients with hypercalcemia are severely dehydrated due to calcium’s effect on the kidney’s ability to concentrate urine (as in nephrogenic diabetes insipidus) and decreased intake. Isotonic saline is used to improve renal blood flow and encourage calcium excretion via exchange for sodium in the renal distal tubule and is the mainstay of treatment. Attention must be paid to volume overload and electrolyte abnormalities. Loop diuretics (furosemide) may be used to help block renal calcium reabsorption while the patient remains volume replete.
Bisphosphonates Most cases of hypercalcemia will require further treatment to decrease bone resorption, specifically bisphosphonates. Bisphosphonates are pyrophosphate analogs that inhibit bone resorption via osteoclasts but do not affect renal tubular absorption. Their effect generally occurs within 36–72 h and lasts from 20–30 days. Osteonecrosis of the jaw is a concerning but rare side effect. More common adverse events include bone pain and therapeutic hypocalcemia.
Typical dosing for pamidronate is 30 mg IV for mild and 60–90 mg IV for severe hypercalcemia infused over 2 h in 50–200 ml of saline or D5W. Pamidronate can be used synergistically with calcitonin. Zoledronate dosing is 4 mg IV over 15 min in 50 ml saline or D5W for most cases of hypercalcemia. Zoledronate should be used with caution in patients with renal insufficiency.
Dialysis Dialysis is available for severe hypercalcemia with acute or chronic renal failure when other treatments cannot be used or are ineffective. However, initiation of dialysis is best reserved for cases in which there are effective treatments for the underlying malignancy.
Supportive Measures and Follow-Up All patients with hypercalcemia should have efforts made toward removal of exogenous calcium (including TPN, supplements, diet), discontinuation of medications that decrease calcium excretion or reduce renal blood flow (like thiazide diuretics, NSAIDs), and increased mobilization or physical activity.
In patients with hypercalcemia, oral phosphate administration can be used to limit oral calcium bioavailability and can make correction of hypercalcemia easier. Usual dosing is 1–2 g/day orally after meals in divided doses but must be monitored in patients with preexisting renal dysfunction or high serum phosphorus levels. Due to potential side effects, intravenous phosphorus is not recommended.
In cases in which no further cancer treatment can be offered, thought should be given to withholding or withdrawing treatment for hypercalcemia. If neurological depression predominates and other symptoms are not significant (anxiety, gastrointestinal distress), withholding treatment may be preferable and should be discussed with the patient and/or the patient’s health-care proxy and family.
Follow-up for hypercalcemia usually includes periodic calcium measurement and bisphosphonate administration.
Second-Line Treatments
Second-line treatments include calcitonin and glucocorticoids. Calcitonin also acts by increasing urinary calcium and can be used synergistically with bisphophonates. Glucocorticoids likely decrease intestinal calcium absorption and inhibit osteoclast-mediated bone resorption and are best used in cases of increased 1, 25(OH)2 vitamin D3 or if the malignancy is responsive to glucocorticoids as a treatment as well (such as lymphoma), given the attendant side effects.
RANK-L (receptor of nuclear factor kappa-B ligand) Agents Denosumab is a fully human monoclonal antibody that targets the RANK ligand. The ligand normally activates pre-osteoclasts into mature osteoclasts and also activates mature osteoclasts. Ongoing clinical trials are testing the efficacy of this agent in hypercalcemia of malignancy.
REFERENCES
1. Cairo M, Bishop M. Tumor lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004; 127: 3–11.
2. Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med. 2011; 364: 1844–1854.
3. Coiffier B, Altman A. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008: 26; 2767–2778.
4. Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000; 342: 1581–1589.
5. Baylis P. The syndrome of inappropriate antidiuretic hormone secretion. Int J Biochem Cell Biol. 2003; 35: 1495–1499.
6. Stewart A. Hypercalcemia associated with cancer. N Engl J Med. 2005; 352: 373–379.