Edwin Choy, Sam S. Yoon, Francis J. Hornicek, Thomas F. DeLaney
SOFT TISSUE SARCOMAS
Soft tissue sarcomas (STS) are uncommon malignancies, arising in about 11,410 persons in the United States each year and accounting for 4390 deaths, mostly due to either locoregional recurrence or distant metastasis (1–4). Although malignant tumors of soft tissue are scarce, benign tumors such as lipomas are 100 times more common. STS occur at any age with a median age of around 50-years old, and are equally common in men and women.
STS constitute a highly heterogeneous group of tumors with respect to anatomical distribution, histologic subtype, and clinical behavior (1). STS occur throughout the body, but nearly one-half occur in the extremities, with about one-third occurring in the lower extremity and 15% occurring in the upper extremity. Another one-third of STS occur in the abdomen, and these are equally divided among intra-abdominal visceral sarcomas (primarily gastrointestinal stromal tumors and leiomyosarcomas) and retroperitoneal sarcomas. Other anatomic sites include the head/neck, trunk, and other miscellaneous sites (e.g., heart).
STS are malignant tumors which arise from the mesodermal tissues (e.g., fat, muscle, connective tissue, and vessels) excluding bone and cartilage. In addition, malignant tumors of peripheral nerve sheaths are usually included despite being ectodermal in origin. There are over 50 different histologic subtypes of STS with the most common being liposarcoma, leiomyosarcoma, fibrosarcoma, and synovial sarcoma. Malignant fibrous histiocytoma was historically the most common subtype but the majority of these are now classified as other subytpes including undifferentiated pleomorphic sarcomas. All suspected STS cases should be reviewed by a pathologist experienced in sarcomas given that about 10% of cases originally designated as STS are in fact not STS and about 20% are initially assigned the incorrect histologic subtype (5). While each histologic subtype may have certain specific clinical behaviors, all STS can generally be categorized into low-, intermediate-, and high-grade tumors. Low-grade tumors grow more slowly, can locally recur after resection, but have a low risk of distant metastases (about 5%). High-grade tumors tend to grow more rapidly, can recur locally, and have the added risk of distant metastasis that can approach 50% for large tumors greater than 5–10 cm in largest dimension.
The treatment of STS has advanced significantly over the past few decades. In particular, evidence has accumulated that in addition to surgery, there are important roles for radiation therapy and chemotherapy in the management of some STS patients. Optimal results from more conservative local treatment strategies require a multidisciplinary approach to the overall management of these patients. The team should include not only an experienced and specialized surgeon, but also a radiation oncologist, medical oncologist, pathologist, and diagnostic radiologist expert in the disease. Additional specialists who may be important in the care of these patients include plastic/reconstructive surgeons, physiatrists working with physical and occupational therapists, psychiatrists, psychologists, and social workers. For this relatively uncommon solid tumor that occurs throughout the body and has over 50 histologic subtypes, evaluation and treatment is best done at a tertiary referral center.
![]() ETIOLOGY
ETIOLOGY
The vast majority of STS occur as sporadic tumors in patients with no identified genetic or environmental risk factors. However, certain genetic syndromes are associated with an increased risk of developing sarcomas including neurofibromatosis 1 (NF1, von Recklinghausen’s disease), hereditary retinoblastoma, and Li-Fraumeni syndrome. Specific genetic abnormalities, evidenced by nonrandom chromosomal aberrations, are well established in certain STS histologic subtypes, and are often utilized in the definitive diagnosis (Table 61-1).
TABLE 61-1 SOFT TISSUE SARCOMA CHROMOSOMAL TRANSLOCATIONS AND GENES INVOLVED

Radiation is recognized as capable of inducing sarcomas of bone and soft tissue. The frequency increases with radiation dose and with the postradiation observation period. Chemotherapeutic agents are likewise associated with risks of sarcoma induction. STS (primarily lymphangiosarcomas) may be observed following massive and quite protracted edema after axillary lymph-adenectomy (Stewart–Treves syndrome). Trauma is rarely a factor in the development of these tumors with the possible exception of desmoid tumors.
![]() STAGING
STAGING
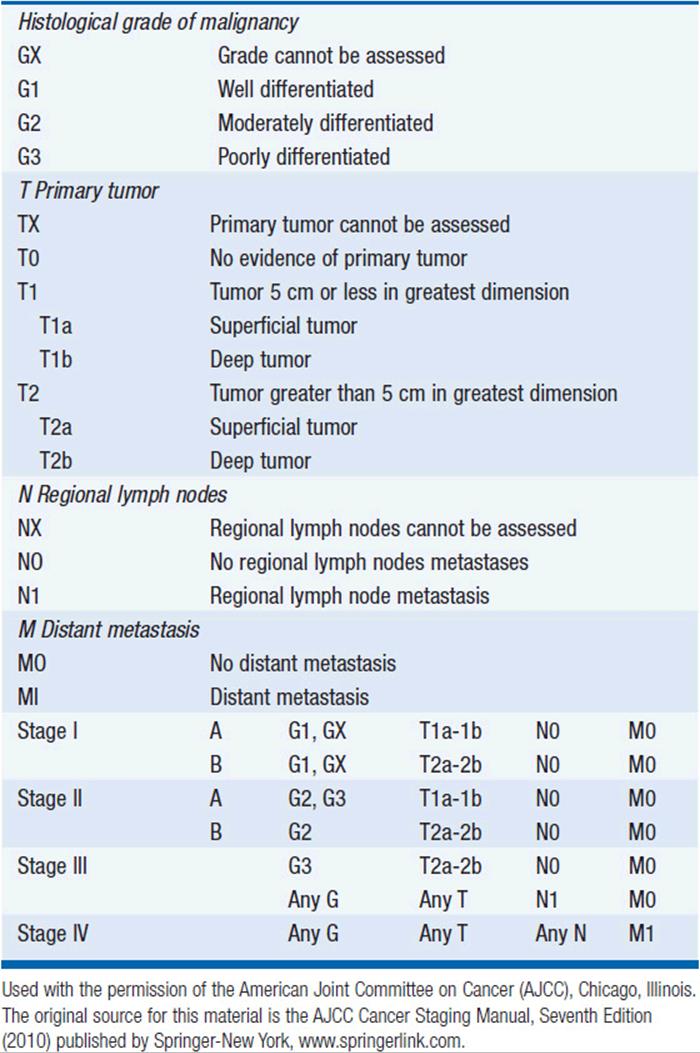
The Task Force on STS of the American Joint Committee on Cancer (AJCC) Staging and End Result Reporting has established a staging system for STS which is an extension of the TNM system to include G for histological grade (Table 61-2). Grade, size, depth, and presence of nodal or distant metastases are the determinants of stage. Of these, grade is particularly important in staging sarcomas. Some institutions will assign grades 1–3, where grade 1 lesions are considered low grade with minimal metastatic potential and the intermediate grade 2 and high grade 3 lesions are considered high grade and capable of metastatic disease. Other institutions use a 2- or 4-tiered system.
TABLE 61-2 AJCC STAGING SYSTEM FOR SOFT TISSUE SARCOMAS
EXTREMITY STS
![]() CLINICAL EVALUATION
CLINICAL EVALUATION
The most frequent initial complaint is that of a painless, enlarging mass for a few weeks to several months. Occasionally, pain or tenderness precedes the detection of a mass. With progressive growth of the tumor, symptoms appear which are usually secondary to infiltration of or pressure on adjacent structures. Interestingly, some high-grade sarcomas in the foot or ankle may have been noticed initially several years prior to diagnosis. One should obtain a complete history and physical examination, with particular attention paid to the region of the primary lesion: definition of size, site of origin (superficial or deep, attached to or fixed to deep structures), involvement or discoloration of overlying skin, functional status of vessels and nerves, mass effect on adjacent organs and joints, and presence of distal edema. Laboratory studies need not go beyond a complete blood count and chemistry panel. There are no tumors markers for STS.
For the primary site, the radiographic evaluation should include a CT scan or MRI. The most useful radiologic study to evaluate an extremity or trunk primary site is the MRI, but CT scans can provide supplemental information. A chest CT should be obtained for high-grade tumors to evaluate for lung metastases. A chest x-ray may be adequate for low-grade tumors. The role of PET scans has yet to be defined, but many primary and metastatic tumors may show increased FDG uptake especially as the grade increases.
An adequate biopsy is required to determine a histologic diagnosis as to tumor type and grade and to determine an optimal treatment strategy. In the majority of cases, the diagnosis can be established by core needle biopsy. Superficial lesions which are readily palpable can be directly biopsied with ultrasound guidance (all lesions should be imaged prior to biopsy), but for tumors which are located at a depth which makes the lesion appear less well defined, a CT-directed approach is advocated. Open biopsies are done less commonly and should be reserved for the uncommon cases where core biopsy is not adequate. The incision for open biopsies should usually be oriented longitudinally such that it can be easily incorporated in the definitive resection. For tumors >3–5 cm in size, and depending on where they are located (i.e., hand or foot are exceptions), an excisional biopsy can sometimes be performed, and incisional biopsies can be performed for larger lesions. Care should be taken to minimize bleeding and contamination of surrounding tissues. Fine needle biopsy can best be employed to confirm metastatic or recurrent tumor when the primary diagnosis is already established.
![]() SURGERY AND RADIATION THERAPY
SURGERY AND RADIATION THERAPY
If STS are “shelled out” as is performed for benign tumors such as lipomas, the local recurrence will be up to 90%. Radical resection of tumors with a margin of normal tissue can decrease the local recurrence rate to 10%–30%. However, many extremity STS grow adjacent to major blood vessels and bones, and the standard operation for many STS up until the early 1980s was amputation. Rosenberg et al. at the National Cancer Institute (NCI) published a randomized trial of amputation versus limb-sparing surgery and radiation (both groups received chemotherapy) in 1982 and demonstrated equivalent overall survival with a local recurrence rate of 0% versus 15% (6). Currently limb-sparing surgery can be performed in over 90% of patients with extremity STS, and overall local recurrence rates are often less than 10%.
Several surgical principles should be followed when resecting STS. First, the preoperative imaging studies should be carefully examined to identify the full extent of tumor penetration as well as the relationship of the tumor to adjacent structures. Second, tumors should be resected with an adequate margin of normal tissue if this can be performed without severe morbidity. The distance is variable and probably can be reduced when high-quality margins such as fascia are present as the border. Tissues such as fat or muscle probably require 1–2 cm of normal tissue because of frequent infiltration of the tumor cells within these tissues. One can often accept a few millimeters of fascia margin but should be more concerned about a close margin of fat or muscle. Third, STS usually do not invade the periadventitial tissue of arteries or the periosteum of bone and can often be dissected along these planes. However, some STS actually arise in the vessels or nerves precluding salvage in terms of resection. Surgeons in general must use considerable judgment in resecting STS and careful discussion with bone and soft tissue radiologists prior to the procedure is important. Positive microscopic margins are very strongly associated with an increased risk of local recurrence, and one should strive for negative microscopic margins in all cases unless this would create major or unacceptable morbidity. In such cases one may rely on adjuvant radiation therapy in order to reduce major surgical morbidity, given radiation can usually be delivered in doses to the extremity that can eradicate microscopic residual disease.
Several studies have defined the essential role of radiation therapy in the local control of STS. Another NCI randomized trial published in 1998 comparing limb-spring surgery alone to surgery and external beam radiation (patients with high-grade tumors all received chemotherapy) demonstrated that radiation reduced local recurrence from 20%–33% to 0%–4% (7). The rate of distant recurrence was the same in both groups. Brachytherapy has also been used to deliver radiation. In a randomized trial of surgery alone versus surgery and brachytherapy, local recurrence for high-grade tumors was reduced from 30% to 5% with brachytherapy. At our institution, brachytherapy is often employed for patients who have a local recurrence after prior surgery and radiation, and this allows the delivery of additional radiation while minimizing morbidity. Brachytherapy or intraoperative radiation therapy can also be used at the time of initial surgery and radiation to deliver a focal boost to areas of close or positive surgical margins.
The order of radiation therapy in relation to surgery is a subject of debate between major sarcoma centers. One randomized trial by the Canadian NCI examined preoperative and postoperative radiation therapy and found no difference in local control (8). Complications were twice as high in the preoperative therapy group (35% vs 17%), but tissue fibrosis and other late complications were more frequent in the postoperative radiation group.
There are certain situations such as difficult anatomical location (e.g., pelvis, spine, or base of skull) or major medical comorbidities (e.g., cardiac dysfunction or metastatic lung cancer) in which a conservative surgical procedure may have much apparent risk and radiation alone is delivered. In one study from Memorial Sloan-Kettering, 25 patients were treated by radiotherapy alone, and local control was achieved in 14 of the 25 patients. To achieve a high probability of local control, higher doses than would be used for residual microscopic disease in the range of 70–75 Gy are essential. Among patients receiving ≥63 Gy, Kepka et al. reported local control in 72% of patients with tumors ≥5 cm in size, 42% for lesions >5 cm but ≤10 cm, and only 25% for lesions >10 cm (9). Because high doses are required for control of these unresected lesions, treatment techniques such as proton beam radiation, or even conventional radiation in an intensity-modulated fashion may be required to deliver dose and stay within the constraints of normal tissue tolerance.
Patients with large (>8–10 cm), deep, high-grade sarcomas present more difficult problems in terms of local control and are at significant risk of distant metastasis. Some groups have combined chemotherapy with surgery and radiation therapy strategies. Eilber and Morton have been proponents of a program, which has consisted of intra-arterial doxorubicin followed by rapid fraction hypofractionated radiation therapy (3.5 Gy/fraction, 28 Gy total) and subsequent local excision. Their data have shown local recurrence rates of <10% with survival rates of 74% in stage III tumors. Among these patients, there was a 5% amputation rate. They have since shown that it is not necessary to provide the doxorubicin by an intra-arterial route. Our institution has employed a regimen of preoperative mesna, Adriamycin, ifosfamide, and dacarbazine (MAID) chemotherapy interdigitated with radiation therapy (44 Gy) followed by postoperative MAID chemotherapy for patients with >8 cm, high-grade STS (10). Five-year local control was 92% and 5-year overall survival was 87%. In experimental protocols, hyperthermic isolated limb perfusion (HILP) with chemotherapeutic agents (e.g., tumor necrosis factor alpha, melphalan, and interferon gamma) has been used to control large tumors that would otherwise require amputation because of proximity to nerve or blood vessels.
![]() ADJUVANT CHEMOTHERAPY
ADJUVANT CHEMOTHERAPY
Although surgery and radiotherapy achieve control of the primary tumor in the majority of patients, many patients (especially those with large, high-grade tumors) develop and die of metastatic disease not evident at diagnosis. Doxorubicin and ifosfamide are the most active chemotherapy agents in metastatic STS. For doxorubicin, objective response rates between 20% and 40% have been reported. Several prospective studies using single-agent doxorubicin failed to show an improvement in disease-free or overall survival in patients receiving postoperative chemotherapy compared with surgery alone. The large EORTC study of adjuvant chemotherapy employed CyVADic consisting of cyclophosphamide, vincristine, doxorubicin, and DTIC found improved 7-year recurrence-free survival (56% vs 43%) but no significant difference in overall survival (63% vs 56%) (11). A meta-analysis of 14 randomized trials of doxorubicin-based adjuvant chemotherapy versus no chemotherapy in STS was performed in 1997 (12) and updated in 2008 with three additional studies using ifosfamide with doxorubicin (13). The adjuvant chemotherapy group had a statistically significant improved rate of distant and overall recurrence (OR was 0.67; 95% CI 0.56–0.82; P = 0.0001), distant recurrence-free survival (70% vs 60%, P = 0.003), but overall survival was only significantly improved in the studies that combined ifosfamide with doxorubicin (OR = 0.56; 95% CI, 0.36–0.85; P = 0.01). However, the toxicities associated with these chemotherapeutic agents dictate caution in generalizing adjuvant chemotherapy to all patients (indeed, different sarcoma centers in North America and Europe have varying degrees of enthusiasm for the use of adjuvant chemotherapy), which argues for and favor continuing enrollment of patients in clinical trials where available.
![]() RETROPERITONEAL STS
RETROPERITONEAL STS
Approximately 10%–15% of soft tissue sarcomas arise in the retroperitoneum. Tumors may be identified on imaging studies for unrelated complaints, or patients may also present with a palpable abdominal mass or with symptoms such as abdominal pain or lower extremity neurologic symptoms. Since the retroperitoneum can accommodate large tumors without symptoms, the average size of tumors in large series is often greater than 10 cm. Upon histologic examination, about two-thirds of tumors are either liposarcomas or leiomyosarcomas, with the remaining tumors distributed among a large variety of other histologic subtypes. Retroperitoneal liposarcomas are further subclassified into well-differentiated/dedifferentiated, myxoid/round cell, and pleomorphic subtypes. Most unifocal tumors in the retroperitoneum that do not arise from adjacent organs will be either benign soft tissue tumors (e.g., schwannomas, paraganglioneuroma, or neurofibroma) or sarcomas. Other malignancies in the differential diagnosis include primary germ cell tumor, metastatic testicular cancer, and lymphoma. Following a careful history and physical examination, radiologic assessment of these tumors is usually performed with an abdominal and pelvic CT scan. Liposarcomas often have a characteristic appearance with large areas of abnormal-appearing fat (well-differentiated liposarcoma) sometimes containing higher-density nodules (dedifferentiated liposarcoma). Patients with high-grade tumors should have a chest CT to evaluate for lung metastases.
The primary treatment for the local control of these tumors is surgical resection (14). The optimal goal of surgical resection is complete gross resection with microscopically negative margins, but this can be difficult to accomplish, and complete gross resection rates in large series are reported to be around 60%. In about three-quarters of cases, complete gross resection requires removal of adjacent viscera. The goal of obtaining negative microscopic margins for large retroperitoneal tumors is frequently not achieved. These tumors are surrounded by a pseudocapsule that often contains microscopic disease, and dissection with a normal tissue margin away from the pseudocapsule is difficult, especially along the posterior aspect of the tumor where it abuts the retroperitoneal fat and musculature. Controversy exists as to the optimal role of radiation therapy for local control of retroperitoneal sarcomas. Those who advocate radiation therapy usually prefer that radiation be delivered preoperatively. With the tumor still in place, normal organs are pushed away from the radiation field, the margin around the tumor at risk of local recurrence is more clearly defined, and the effective radiation dose required to control microscopic disease is likely lower.
In the extremity, the local control of sarcomas treated with total gross resection with positive microscopic margin and adjuvant radiation therapy is about 75%. Typically, positive microscopic margins are treated with a boost of intraoperative, perioperative brachytherapy, or additional postoperative radiation to a total dose of about 60–70 Gy, although some recent data suggest that selected patients such as those with well-differentiated liposarcoma and a focal, planned positive margin following preoperative radiation may not benefit additional postoperative boost radiation (15). It is reasonable to assume that total gross resection of retroperitoneal tumors along with adequate doses of radiation could achieve local control rates similar to that seen for extremity tumors resected with positive microscopic margins. However, unlike the extremity, it is difficult to deliver high doses of radiation to the abdomen. The availability of intensity-modulated radiation therapy, proton beam radiation, and intraoperative radiation therapy may facilitate the efficacy and minimize morbidity of adjuvant radiation therapy for these tumors. In a report from our institution, 29 patients were treated with preoperative radiation to a median dose of 45 Gy and then underwent complete gross resection (16). Intraoperative radiation therapy (IORT) (10–20 Gy) was delivered to 16 of the 29 patients. Local control at 5 years was 83% for patients who received both preoperative and intraoperative radiation therapy and 61% for those who received only preoperative radiation. More recently, we have incorporated the use of preoperative proton beam radiation therapy and/or IMRT along with aggressive anterior surgical resection +/− and intraoperative radiation therapy for retroperitoneal tumors to further minimize to maintain high rates of local tumor control while minimizing morbidity on adjacent structures. In a report of this experience with a median follow-up of 33 months, Yoon et al noted only two local recurrences among 20 patients treated for primary retroperitoneal sarcomas. Among all 28 patients in the series, 28% had surgical complications and 14% had radiation related complications (17).
![]() FOLLOW-UP
FOLLOW-UP
The intensity of follow-up visits and imaging studies varies between institutions, and can also be varied according to tumor grade. The National Comprehensive Cancer Network (NCCN) published guidelines in 2011 suggesting for low-grade extremity tumors, patients should be evaluated by history and physical examination every 3–6 months for the first 2–3 years, then annually. Chest x-rays should be obtained every 6–12 months. Imaging of the primary tumor site depends on the location and the risk of locoregional recurrence. For superficial tumors, physical examination is often sufficient. For deeper extremity tumors, CT scan or MRI may be performed.
For high-grade lesions, the NCCN guidelines suggest a history a physical examination every 3–6 months for 2–3 years, then every 6 months for the next 2 years, then annually. Chest CT scans or chest x-rays should be obtained at each visit.
METASTATIC DISEASE
While local control of STS can be attained in >90% of patients, up to 50% of patients present with or develop metastatic disease. Median survival after the development of metastatic disease is 8–12 months, although a sizeable minority of patients can live several years if the disease is indolent. The most common site of metastatic disease for extremity and trunk STS is the lung. Intra-abdominal and retroperitoneal sarcomas metastasize with about equal frequency to the lung and liver. STS rarely metastasizes to regional lymph nodes (about 5%) except for certain histologic subtypes including clear cell sarcoma, epithelioid sarcoma, rhabdomyosarcoma, hemangiosarcoma, and synovial sarcoma.
As noted above, doxorubicin and ifosfamide have been demonstrated to be the most active chemotherapy agents in widely disseminated soft tissue sarcoma (18). Response rates range from about 20% to 30%, and there is some evidence that higher doses can result in increased response rates. These two agents carry significant risks of toxicity: doxorubicin dosage is limited by cardiotoxicity and ifosfamide causes hemorrhagic cystitis and nephrotoxicity. Ifosfamide-induced hemorrhagic cystitis can be avoided by adding the protective agent mesna. Another agent with some activity against STS is dacarbazine (DTIC), with reported response rates around 20%. However for metastatic disease, complete responses are uncommon (<15% even with combination therapy), duration of response averages 8 months, and as yet the higher response rates seen with intense combination therapy have not been proven to provide a survival advantage over that conferred by sequential single agent treatment.
A variety of combination chemotherapy regimens for metastatic disease have been studied in randomized clinical trials. Most of these studies were small and included mixed histologies (see Table 61-3). Most of these trials include doxorubicin (or epirubicin) and an alkylating agent. Overall survival for any combination chemotherapy regimen has not been clearly demonstrated to be superior to doxorubicin alone, but trials have been underpowered due to the rarity of this disease type. A commonly used combination chemotherapy regimen is mesna, adriamycin, ifosfamide, and dacarbazine (MAID). In a large phase II trial, this regimen achieved an overall response rate of 47%. When MAID was compared to the combination of adriamycin and dacarbazine in a randomized trial, the response rate was higher for MAID (32% vs 17%). However, there were more toxic deaths in the MAID group and there was no survival advantage to MAID. Higher doses of chemotherapy can be given with better control of toxicity if granulocyte-colony-stimulating factor (G-CSF) is used.
TABLE 61-3 SELECTED RANDOMIZED CHEMOTHERAPY TRIALS FOR METASTATIC SOFT TISSUE SARCOMAS

Other trials evaluating the use of other chemotherapy agents such as docetaxel, gemcitabine, vinorelbine, and topotecan all show modest rates of significant tumor shrinkage. Some histologies may be particularly sensitive to specific agents, such as scalp angiosarcoma to paclitaxel. Another agent that has shown some promise in recent trials against STS is Ecteinascidin 743 (ET-743), which is also known as Yondelis or trabectedin. This drug seems to have particular activity against myxoid liposarcomas and is commercially available in Europe, Asia, and Australia, but it is not yet approved for use in the United States. Patients with metastatic disease should be strongly considered for enrollment in investigational or experimental trials.
Two randomized phase III studies in patients with advanced sarcomas have been performed to demonstrate improvement in progression-free survival. The PALETTE trial randomized 372 patients with metastatic soft tissue sarcoma in 2:1 fashion to receive pazopanib (a multiple tyrosine kinase inhibitor) or placebo (19). The median progression-free survival time was 4.6 months for pazopanib and 1.6 months for placebo. The SUCCEED trial took 711 patients with metastatic sarcoma who experienced stable disease or better to standard chemotherapy to be randomized in 1:1 fashion to either ridaforolimus (an mTor inhibitor) or placebo (20). The median progression-free survival time was 17.7 weeks for ridaforolimus and 14.6 weeks for placebo. This was found to be statistically significant. However, neither trial demonstrated improvement in overall survival. Based on these results, pazopanib was approved for marketing to patients with sarcomas by the FDA. Ridaforolimus was not recommended for FDA approval.
There are currently five other randomized, phase III studies underway at the time this manuscript is being prepared. These include a first-line study of: (1) trabectedin versus doxorubicin, (2) doxorubicin with or without TH-302, (3) doxorubicin with or without palifosfamide, and a third-line study of (4) eribulin versus dacarbazine and (5) trabectedin versus dacarbazine. These studies are described in clinicaltrials.gov.
Surgical resection has been performed for isolated STS metastases to the lung or liver. For select patients with STS metastases isolated to the lung, surgical resection (a.k.a. metastasectomy) can be performed, and 5-year survival can approach 20%–30%. Intra-abdominal and retroperitoneal STS metastasize commonly to the liver as well as the lung. Isolated, resectable liver metastases from STS are less common than isolated, resectable lung metastases, but several small series have examined partial hepatectomy for these patients and 5-year survival can reach 30%.
GASTROINTESTINAL STROMAL TUMORS
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumor of the gastrointestinal tract and originate from the interstitial cells of Cajal (15). Interstitial cells of Cajal and the vast majority of GIST express c-KIT, which is a 145-kD transmembrane glycoprotein that acts as the receptor for stem cell factor (SCF). Prior to the identification and availability of immunohistochemistry for c-KIT, the majority of GIST were thought to be of smooth muscle origin and termed leiomyomas, leiomyosarcomas, and leiomyoblastomas. It is now known that the majority of stromal tumors of the gastrointestinal tract are GIST. Ninety-five percent of GIST stain positive on immunohistochemistry for c-KIT and the majority of GIST have a mutation in the c-KIT gene. Unlike most other cancers, GISTs seem highly dependent on this single pathway for neoplastic growth.
GIST can occur at nearly all ages, with the median age being around 60. The incidence is roughly equal in men and women. There are roughly 5000 cases per year in the United States. GISTs are located most commonly in the stomach (60%) followed by the small intestine (30%), colon/rectum (5%), and esophagus (5%). These tumors can be found on upper endoscopy, where they appear as submucosal lesions. However, even large GIST of the stomach may not be seen on endoscopy if they are pedunculated or exophytic. Endoscopic biopsy can establish the diagnosis, especially following staining for c-KIT expression. For small bowel tumors, CT-guided biopsy poses the risk of inadequate tissue and spillage of tumor cells and is likely not necessary given most isolated small bowel tumors require resection. Staging workup should include an abdomen and pelvis CT scan to rule out intra-abdominal or liver metastases.
GIST can range from small, pedunculated lesions to large lesions with adherence or invasion of surrounding tissues and organs. Thus the surgical approach can be quite varied but certain principles should be followed. Upon surgical exploration, the liver and peritoneal cavity should be examined for possible metastatic disease. GISTs uncommonly metastasize to lymph nodes so a regional lymphadenectomy is not required. Thus for a pedunculated gastric tumor, wedge resection of the gastric wall along with resection of the tumor is adequate. Some gastric tumors encompass a large portion of the stomach and a formal distal, subtotal, or even total gastrectomy may be required. For small bowel and colon tumors, segmental bowel resection can be performed. Small rectal GIST can be removed through transanal procedures while larger tumors may require low anterior resection or even abdominal-perineal resection, although there are reports of excellent responses using neoadjuvant imatinib and radiation as a means of allowing anal sphincter sparing surgery (21). Some large GISTs with significant necrosis are susceptible to tumor rupture and spillage, which can lead to intraperitoneal spread of disease, and so these tumors should be manipulated carefully. For large GISTs that invade adjacent organs, an en bloc resection of the entire tumor mass should be performed.
Risk of recurrence is most closely related to mitotic rate, size, and location. Table 61-4 shows the estimated risk of recurrence for resected GIST based on these three parameters, mitotic rate, tumor size, and tumor location (22). The most common sites of metastasis are the liver and peritoneal cavity, and less common sites include the lung and bone. Prior to the introduction of imatinib, GISTs were found to be highly resistant to chemotherapeutic agents. Imatinib inhibits both the c-KIT receptor and platelet-derived growth factor receptor (PDGFR) tyrosine kinases. Several large studies have shown imatinib to be highly effective against metastatic GIST, with a partial response rate of about 40% and stable disease rate of about 30% (see Table 61-5). The median time to progression is 19–24 months. Even tumors that are sensitive to imatinib may take months to show a decrease in tumor size on CT scans, and so PET scans have been employed to assess response. GISTs are usually positive on PET scans, and response to imatinib as demonstrated by decrease in PET activity can frequently be seen immediately after initiation of therapy. When resistance to imatinib ultimately develops, the dose can be escalated or other new targeted biologic agents may be considered. The most studied agents include sunitinib, sorafenib, and regorafenib.
TABLE 61-4 PERCENT OF PATIENTS WITH PROGRESSIVE DISEASE BASED ON MITOTIC RATE, SIZE, AND LOCATION

TABLE 61-5 IMATINIB TRIALS FOR METASTATIC GIST

The role of surgery for metastatic GIST has yet to be defined. Some investigators surgically resect or debulk imatinib-responsive disease to decrease tumor burden and hypothetically delay the development of imatinib resistance. When resistance to imatinib ultimately develops, the dose can be escalated or other newly targeted biologic agents, which have shown promise for imatinib-resistant GIST, may be considered. The most studied agent is sunitinib, which targets not only c-KIT and PDGFR but also the VEGF receptors, fms-related tyrosine kinase (Flt3), and the Ret oncogene. In a randomized study of 312 patients with imatinib-refractory GIST, sunitinib achieved stable disease for 27.3 weeks while placebo achieved stable disease for 6.4 weeks (23). Sunitinib was FDA approved for second-line therapy of metastatic GIST on January 2006, and Regorafenib was FDA approved for third-line therapy of metastatic GIST on February 2013.
DESMOID TUMOR (AGGRESSIVE FIBROMATOSIS)
Desmoid tumors are benign but locally infiltrative neoplasms arising from fibroblastic stromal elements that can grow at variable rates. Although neo-plastic and locally aggressive, desmoids do not have the capacity to establish metastatic lesions. Desmoid tumors are uncommon (possibly about 1000 per year in the United States), slightly more common in females than males, and occur predominantly in individuals of 15–60 years of age. There is no significant racial or ethnic distribution. The etiology of desmoid tumors is not known, but these tumors occur commonly in patients with Gardner’s syndrome and occur more frequently at sites of trauma or surgery.
Desmoid tumors usually present as a painless or minimally painful mass with a history of slow growth, but can cause symptoms if they impinge on adjacent structures. Desmoid tumors occur at virtually all body sites; most commonly in the torso (shoulder girdle and hip-buttock region) and the proximal aspect of extremities. The location is usually deep in the muscles or along fascial planes. Multiple lesions at distant sites are infrequent; however, additional lesions are not rare on the same extremity following the initial treatment.
The natural history of desmoids can be quite variable. They commonly grow relatively slowly, with periods of comparative stability or even in certain cases temporary regression. Desmoid tumors can be locally progressive with infiltration of adjacent normal tissues and structures. They may become locally malignant and highly destructive of normal tissue leading to the death of the patient. Spontaneous regressions have been observed, and regrowth is not observed in all patients following grossly incomplete surgical resection.
Desmoid tumors may be treated by surgical resection with a wide margin when medically and technically feasible. In recent years, the option to observe initially asymptomatic lesions has also emerged as an appropriate management strategy, with the finding that that approximately 50% of patients with desmoid tumors exhibit spontaneous growth arrest during simple follow-up with either medical therapy only or no therapy at all (24, 25). Since these tumors are benign, treatments with potentially serious late sequelae should be avoided if at all possible. Radiation therapy is an effective option for patients who are not good surgical candidates or decline surgery and, as an adjunctive therapy, for patients with grossly or microscopically positive margins or for those patients with recurrent disease. While some advocate adjuvant radiation for primary disease, our institution generally reserves radiation for patients with recurrent tumors after prior surgery or unresectable tumors that have progressed after observation or simple systemic treatment such as nonsteroidal anti-inflammatory drugs (NSAIDs) because this is a benign disease. Results from a number of centers demonstrate that radiation alone (50 Gy) or radiation combined with surgery in patients with positive margins achieves permanent control of desmoid tumors in approximately 70%–80% of patients (26). We also tend to try to defer the use of radiation as long as possible in pediatric patients, preferring to use any of the multiple systemic agents prior to radiotherapy in the child, in whom radiation may be associated with growth arrest, soft tissue atrophy, and late second malignancies.
There is increasing evidence for a role for systemic therapy in patients whose desmoid tumors cannot be controlled without morbid surgery and/or radiation (27). Increasing experience is accumulating with systemic agents in patients with advanced or recurrent disease, especially at intra-abdominal and abdominal wall sites. There are, for example, a number of anecdotal reports of excellent response to tamoxifen, the antiestrogen toremifine, and progestational agents. There are also documented responses to NSAIDs (most often sulindac) alone or in combination with tamoxifen. Regression is usually partial and may take many months after an initial period of tumor enlargement. Low-dose chemotherapy based upon methotrexate and vinblastine has obtained worthwhile response rates in patients with desmoid tumors, particularly in children. The combination of methotrexate and vinorelbine may produce a similar response to methotrexate and vinblastine with less neurotoxicity. In addition, there are anecdotal reports of doxorubicin-based drug protocols with good responses. For patients with Gardner’s syndrome and intra-abdominal desmoid tumor, one should often avoid aggressive surgery given the relatively poor results of surgery and the higher rate of recurrence. Such patients can be treated with tamoxifen, NSAIDs, or low-dose chemotherapy. Surgery is the second option if there is no response.
BONE SARCOMAS
Malignant tumors of the skeletal system are rare. There were an estimated 3010 new bone sarcomas cases in the United States in 2013 with 1440 deaths. The most common primary bone sarcoma is osteosarcoma, followed by chondrosarcoma and Ewing’s sarcoma. Osteosarcoma and Ewing’s sarcoma occur primarily in childhood and adolescence. Other primary bone sarcomas, including chondrosarcomas, fibrosarcoma, and MFH, occur primarily in adults. Malignant tumors of bone must be differentiated from benign bone and cartilage tumors such as osteochondroma, enchondroma, osteoid osteoma, osteoblastoma, and desmoplastic fibroma, and giant cell tumor of bone and other malignant tumors such as myeloma and metastatic carcinoma. Just as for soft tissue sarcomas, the NCCN has been able to provide guidelines for management.
![]() OSTEOSARCOMA
OSTEOSARCOMA
Osteosarcoma is the most common malignant bone tumor (28). Tumors are usually located in the metaphysis of long bone, especially the distal femur, proximal tibia, and proximal humerus. The most common presentation of patients with bone sarcomas is pain or swelling in a bone or near a joint. At the time of presentation, 10%–20% of patients have macroscopic metastatic disease and over 80% of patients likely harbor micrometastatic disease. Osteosarcomas metastasize primarily to the lung (90%) and less commonly to other bone sites (10%). Rarely do they metastasize to lymph nodes except in advanced stages of the disease. On pathologic analysis, the characteristic feature of osteosarcoma is the presence of tumor cell-produced osteoid. Several histologic subtypes of osteosarcomas exist: conventional (including osteoblastic, chondroblastic, and fibroblastic), small cell, radiation induced, sclerosing, extra-osseus, telangiectatic (high-grade aggressive with blood-filled cavities), and juxtacortical (parosteal [low grade] and periosteal [usually intermediate to high grade with cartilage]). The low-grade tumors have a much better prognosis than intermediate- to high-grade tumors, with the former not requiring chemotherapy. Most osteosarcomas are conventional approaching 80% of the total number per year.
Diagnosis and Staging
The radiologic appearance of osteosarcomas and other malignant bone tumors are usually different from that of benign tumors. Benign tumors usually have well-circumscribed borders with no cortical destruction or periosteal reaction. Malignant tumors often have irregular borders, bone destruction, periosteal reaction, and soft tissue extension. Patients should be fully evaluated by history and physical examination, plain radiographs, MRI and CT scan of the primary tumor site, chest CT, and bone scan. The diagnosis can usually be established by CT-guided core biopsy or open incisional biopsy. Often there exists an associated soft tissue component that can be biopsied for presence of most aggressive cells and it avoids producing a hole in the bone. The most commonly used staging system was developed by Enneking et al. and is outlined in Table 61-6 (29).
TABLE 61-6 MUSCULOSKELETAL TUMOR SOCIETY STAGING SYSTEM FOR BONE SARCOMAS

Surgery and Radiation
A wide surgical margin is preferred for resection of tumor coordinated with chemotherapy in intermediate- to high-grade osteosarcomas. In the axial locations where tumors tend to be larger when discovered the outcome in general is worse. In the extremities, about 90% of osteosarcomas can be removed using limb-sparing surgery, although amputation is sometimes necessary to achieve a negative surgical margin. Limb-sparing surgery is divided into three components:
1. Resection of tumor: Tumors are ideally removed with a rim of normal tissues to obtain a negative gross and microscopic margin. For low-grade lesions in which patients do not get chemotherapy, a marginal margin through the reactive zone may allow for salvage of those neurovascular structure abutting the tumor without jeopardizing local or distant disease.
2. Skeletal reconstruction: The skeletal defect is usually reconstructed but not always, especially if the tumor is located in an expendable bone like the proximal fibula. Most defects created by the resection can be reconstructed using a variety of techniques including autologous bone graft, allograft, endoprosthesis, and rotationplasty.
3. Muscle and soft tissue coverage: Local muscle, fat, and skin can be mobilized to cover the tumor resection bed and skeletal reconstruction. For larger defects, rotational or free flaps may be required. Areas more prone to infection like the proximal tibia may routinely get a gastrocnemius muscle flap.
Osteosarcomas are relatively resistant to radiation. Radiation therapy is generally not used in the primary treatment of most patients osteosarcomas, but is reserved for patients who refuse definitive surgery, have no good surgical option (i.e., base of skull), have grossly or microscopically positive resection margins, have less than wide resection margins and poor histologic response, have primary lesions in sites with high rates of relapse after surgery including head and neck, spine, or pelvis, present with a pathologic fracture and might be at higher risk of local recurrence, or require palliation. Retrospective reports suggest potential benefit for the addition of radiation therapy in these settings (30). For osteosarcomas in patients with extremity lesions who refused amputation, a combination of chemotherapy and primary radiation therapy (median dose 60 Gy) has been reported to obtain a 5-year local control of 56% in 31 patients; local control was 11/11 in selected patients who had a good response based on imaging and normalization of alkaline phosphatase. However, at our institution, surgical resections of osteosarcomas even in difficult locations requiring internal hemipelvectomy have been performed with acceptable morbidity, and local recurrence risk is substantially decreased. For patients for whom resection has been incomplete (43 patients) or has not been done (12 patients) because no good resection option exists (such as skull base) or in whom resection at such difficult locations as the upper sacrum has been declined, high-dose proton-based radiation or chemoradiation has provided durable local control for 73% of patients in one recent series (31).
Chemotherapy
In the 1970s prior to the use of adjuvant chemotherapy, <20% of osteosarcoma patients treated with surgery alone lived over 5 years. With the introduction of adjuvant chemotherapy (high-dose methotrexate), 5-year survival rates rose to 40%–60%. Prospective randomized trials performed in the 1980s confirmed the utility of adjuvant chemotherapy. Effective agents include high-dose methotrexate, doxorubicin, bleomycin, cyclophosphamide, dactinomycin, vincristine, cisplatin, ifosfamine, and etoposide (Pediatric Oncology Group, POG 8651). Neoadjuvant chemotherapy was originally introduced at Memorial Sloan-Kettering in concert with increased utilization of limb-sparing surgery. Chemotherapy was delivered to patients prior to surgery while awaiting creation of custom prosthetics, and retrospective analysis suggested these patients fared better than those that received postoperative chemotherapy alone. A randomized trial of comparing neoadjuvant and postoperative chemotherapy versus purely postoperative chemotherapy that consisted of alternating courses of high-dose methotrexate with leucovorin rescue, cisplatin and doxorubicin, and bleomycin, cyclophosphamide, and dactomycin given before or after surgical resection showed equivalent 5-year relapse-free survival (61% vs 65%) and limb salvage rates (50% vs 55%).
Currently about 65% of osteosarcoma patients treated with adjuvant chemotherapy attain long-term survival. Most centers deliver chemotherapy in the neoadjuvant setting, and this has several theoretical advantages: (1) early delivery of treatment for micrometastases, (2) increase in limb-salvage rates, and (3) assessment of tumor response. Tumor response to neoadjuvant chemotherapy (>90% tumor necrosis) is the most important prognostic factor for survival, and poor responders can be switched to alternative postoperative chemotherapy regimens. There exists some controversy as to the benefits of switching to alternative chemotherapy regimens in patients with <90% tumor necrosis.
The optimal chemotherapy regimen is still controversial. Many centers offer children, adolescents, and younger adults treatment similar to the POG regimen described above in the neoadjuvant setting. Ifosfamide and etoposide are used in some regimens. Older adults may not tolerate doxorubicin and cisplatin for more than 6 cycles. However, high-dose methotrexate and ifosfamide can also be used for most adults.
Recurrent and Metastatic Disease
Patients with metastatic osteosarcoma have long-term survival ranging from 10% to 40% with chemotherapy and surgery and in some cases radiation therapy. Patients with isolated lung metastases have a more favorable prognosis than those with bone metastases. The most active single agents are high-dose methotrexate, doxorubicin, cisplatin, and ifosfamide, with response rates ranging between 20% and 40%, and a variety of combination regimens have been used. Patients who recur following initial treatment with adjuvant chemotherapy and surgery tend to have chemotherapy-resistant disease. For isolated lung metastases that are few in number and do not invade the pleura, complete resection of all lesions can result in 5-year survival up to 35%. Unresectable metastatic disease can be treated with alternative chemotherapy regimens. Most patients who have already received doxorubicin and cisplatin (with or without high-dose methotrexate) can receive etoposide and ifosfamide with or without carboplatin, and such patients should be considered for enrollment in clinical trials.
![]() EWING’S SARCOMA FAMILY OF TUMORS
EWING’S SARCOMA FAMILY OF TUMORS
Ewing’s sarcoma was initially described by James Ewing as a tumor that was responsive to radiation treatment, and is rare tumors that arise in bone and less commonly in soft tissue. There is currently a spectrum of neoplastic diseases known as the Ewing’s sarcoma family of tumors, which includes Ewing’s sarcoma, primitive neuroectodermal tumors (PNET), adult neuroblastoma, malignant small cell tumor of the thoracopulmonary region (Askin’s tumor), paravertebral small cell tumor, and atypical Ewing’s sarcoma. These tumors are thought to be derived from a common cell of origin, share pathologic characteristics, and often have common chromosomal translocations (Table 61-1). As with osteosarcomas, these tumors primarily occur in adolescents and young adults and present with pain or swelling in a bone or joint. They usually involve flat bones or the metaphyseal and diaphyseal regions of tubular bones. While less than 25% of patients present with overt metastases, the majority of patients likely harbor micrometastatic disease, and the common sites of metastasis are the lung, and bone marrow. Workup and diagnosis are performed in a way similar to that for patients with osteosarcoma, except that bone marrow biopsy may be useful.
The surgical principles used for these tumors are similar to those for osteosarcoma. In contrast to osteosarcoma, the soft tissue mass present with Ewing’s sarcoma usually shrinks with preoperative chemotherapy. In osteosarcoma the soft tissue mass will frequently demonstrate changes radiographically but not necessarily in the size of the mass. Deciding whether to use radiation or surgery for local control in the pelvis and axial regions is not always clear. Some groups have even proposed a combination therapy for management of these cases. In general, Ewing’s sarcoma/PNET is relatively sensitive to radiation which is often applied to patients with positive surgical margins and for primary radiation local therapy in cases where surgery would be highly morbid. Selection factors make it difficult to determine whether primary radiotherapy results in as good local control as surgery alone or as surgery combined with radiation therapy. The major concern with the use of radiation therapy is the potential for late radiation-induced malignancies, particularly in younger patients. Radiation therapy has also been used in patients with positive surgical margins, although it is generally preferred to avoid surgery if positive margins are anticipated, because the dose generally applied for positive margins (50.4 Gy) is not much lower than that employed for primary radiotherapy alone (55.8 Gy) and the use of radiation therapy alone in this setting is generally expected to result in less morbidity than both surgery and radiotherapy.
For Ewing’s sarcoma/PNET, standard chemotherapy includes vincristine, doxorubicin, and cyclophosphamide with or without actinomycin D (VDCA or VDC) alternating with ifosfamide and etoposide (IE). Most current protocols employ 4–6 cycles of chemotherapy over 12 weeks. Usually at least a few of the cycles are given as neoadjuvant chemotherapy preoperatively prior to local treatment with surgery or radiotherapy followed by additional chemotherapy that ultimately approaches close to a year of treatment. With modern multimodality treatment, long-term survival can be achieved in 70%–80% of patients with nonmetastatic disease.
For patients with metastatic disease, high-dose chemotherapy with or without whole body radiation and autologous hematopoietic stem cell support has been used. Patients with metastatic disease have a better prognosis when the lung is involved compared to bone and bone marrow. With chemotherapy, 5-year survival can be 20%–40%. Low-dose bilateral lung radiation can also be used, and isolated lesions in other locations can be controlled with radiation.
![]() CHONDROSARCOMAS
CHONDROSARCOMAS
Chondrosarcomas are the second most common malignant bone tumor and occur in patients usually in their third to fifth decade of life (32). The most common anatomic locations are the pelvis (31%), femur (21%), and shoulder girdle (13%). They occur in five primary types: 75% are “conventional” chondrosarcomas of either central (arising within a bone) or peripheral (arising from a bone surface) types and 25% are chondrosarcoma variants (mesenchymal, differentiated, and clear cell). The central and peripheral types of chondrosarcoma can be primary tumors or arise secondary to an underlying neoplasm such as a benign cartilage tumors. Most chondrosarcomas are grade I or II and uncommonly metastasize, but grade III lesions have the same metastatic potential as osteosarcomas. In a large single institution series of 344 chondrosarcomas, overall 5-year survival was 77% (33). Local recurrence developed in 20% and distant metastases in 14%. Local recurrence was higher for shoulder and pelvis tumors, high-grade tumors, and tumors with intralesional or marginal resections. High-grade was also an important prognostic factor distant recurrence. These results are similar to those reported by our institution (34).
The primary treatment of chondrosarcomas is surgical resection. These tumors are highly resistant to chemotherapeutic agents, possibly due to their extracellular matrix. Intralesional excision of grade I tumors has been advocated due to their benign behavior, although tumors located in the spine and pelvis can have a more aggressive biology. Grades II and III tumors should be resected with a wide surgical margin. Although it has been stated repeatedly that chondrosarcoma is a radioresistant tumor, several recent reports have demonstrated the effectiveness of conventional radiation therapy for this histology and it has been successfully employed in patients considered to be a high risk for local recurrence such as those with lesions located at complex sites where complete resection would be anticipated to be a problem such as spine, sacral, cranial or skull-base chondrosarcoma, and/or lesions with close or involved surgical margins and/or high-grade lesions (35). High rates of local control of these lesions in the base of skull/cervical spine and lower spine and sacrum with proton radiation therapy have been reported. The indications for radiation therapy for chondrosarcomas include unresectable or subtotally tumors, those resected with positive margins, and recurrent tumors. Because of the need for high radiation doses for control of these tumors and the proximity of radiosensitive critical structures in the skull base, spine, and pelvis, proton beam radiation, which has no exit dose beyond the target, is advantageous for treatment of tumors in these sites (36, 37).
There is no standard chemotherapy for chondrosarcoma, but patients with metastatic disease should be considered for enrollment in clinical trials.
CHORDOMAS
Chordomas are rare tumors that arise from the notochordal remnant in the midline of the neural axis (20). They occur most commonly in the sacrococcygeal region (50%) and base of skull (35%), but can occur in the cervical, thoracic, and lumbar spine regions (15%). Extraxial or parachordomas have been reported in unusual anatomic locations like the tibia and wrist. Sacrococcygeal lesions usually present with local pain. Patient can also have constipation or urinary symptoms. Base of skull lesions usually present with headache, visual changes, or cranial nerve dysfunction. CT scans and MRI are used for radiologic evaluation. CT imaging of the liver and lung should be performed to rule out metastases, and CT-guided biopsy usually establishes the diagnosis.
These tumors do metastasize in about 30% of patients with sacrococcygeal lesions (38) and a lower percentage of patients with skull base lesions, they present formidable local tumor control challenge and the tumors can be lethal due to their location adjacent to vital structures, locally aggressive behavior, and high rate of recurrence. Local control is difficult and local recurrence and complications often lead to death. The best chance at local control for these tumors is with the initial treatment, and such treatment should include aggressive surgical resection if feasible and radiation therapy when extraosseous extension is present. Even in small lesions after radical resection, recurrence rates are as high as 50%–100%, with local control and survival curves following a continuous downward slope in many series. Salvage treatment after local failure rarely is curative. A possible dose-response relationship for conventional radiation treatment has been reported. One of the best results of conventional, postoperative radiation treatment was published by Keisch et al (39). The rate of actuarial disease-free survival at 5 years was significantly better for patients undergoing surgery and radiotherapy (60%) compared to that of patients undergoing surgery only (25%), although patients continued to recur beyond 5 years. Investigators at the Lawrence Berkeley Laboratory (LBL) and our institution have demonstrated promising results for chordomas at the base of skull and cervical spine using charged particle irradiation, although local control is less favorable than with chondrosarcomas. Munzenrider and Liebsch reported that the 10-year local control for skull base tumors was highest for chondrosarcomas, intermediate for chordomas in males, and lowest for chordomas in females (94%, 65%, and 42%, respectively). For cervical spine tumors, 10-year local control rates were not significantly different for chordomas and chondrosarcomas (54% and 48%, respectively), nor was there any significant difference in local control between males and females. Hug et al. reported 5-year rates of local control and survival for lower spine and sacral chordomas of 53% and 50%, respectively, with a mean dose of 74.6 CGE (cobalt gray equivalent). DeLaney et al noted no local recurrences among 23 patients with primary spine chordomas treated with high-dose proton-based radiation (70.2 Gy for microscopic disease and 77.4 Gy for gross disease) with surgery in the majority of patients, although similar treatment resulted in local tumor control in only 3/6 patients with local tumor recurrence after prior surgery A trend for improved local control was noted for primary lesions compared to recurrent tumors, with radiation doses of at least 77 CGE and less residual tumor burden.
There is no standard chemotherapy for chordomas, but for metastatic disease there is some evidence for the use of imatinib, and patients should be considered for enrollment in clinical trials.
REFERENCES
1. Brennan MF, Lewis JL. Diagnosis and Management of Soft Tissue Sarcoma. Martin Dunitz, London, 2002.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63: 11–30.
3. http://www.cancer.gov/cancertopics/pdq/treatment/adult-soft-tissue-sarcoma/HealthProfessional. Accessed August 20, 2013.
4. Lewis JJ, Leung D, Woodruff JM, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg. 1998; 228: 355–365.
5. Lurkin A, Ducimetière F, Vince DR, et al. Epidemiological evaluation of concordance between initial diagnosis and central pathology review in a comprehensive and prospective series of sarcoma patients in the Rhone-Alpes region. BMC Cancer. 2010; 10: 150.
6. Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg. 1982; 196: 305–315.
7. Yang JC, Chang AE, Baker AR, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol. 1998; 16: 197–203.
8. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002; 359: 2235–2241.
9. Kepka L, DeLaney TF, Suit HD, et al. Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys. 2005; 63:852–859.
10. DeLaney TF, Spiro IJ, Suit HD, et al. Neoadjuvant chemotherapy and radiotherapy for large extremity soft-tissue sarcomas. Int J Radiat Oncol Biol Phys. 2003; 56: 1117–1127.
11. Bramwell V, Rouesse J, Steward W, et al. Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma-reduced local recurrence but no improvement in survival: a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1994; 12: 1137–1149.
12. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997; 350: 1647–1654.
13. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008; 113: 573–581.
14. Lewis JJ, Leung D, Woodruff JM, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg. 1998; 228: 355–365.
15. Al Yami A, Griffin AM, Ferguson PC, et al. Positive surgical margins in soft tissue sarcoma treated with pre-operative radiation: is a postoperative boost necessary? Int J Rad Oncol Bio Phys. 2010; 77: 1191–1197.
16. Gieschen HL, Spiro IJ, Suit HD, et al. Long-term results of intraoperative electron beam radiotherapy for primary and recurrent retroperitoneal soft tissue sarcoma. Int J Radiat Oncol Biol Phys. 2001; 50: 127–131.
17. Yoon SS, Chen YL, Kirsch DG, et al. Proton-beam, intensity-modulated, and/or intraoperative electron radiation therapy combined with aggressive anterior surgical resection for retroperitoneal sarcomas. Ann Surg Oncol. 2010; 17: 1515–1529.
18. Brennan MF, Alektiar KM, Maki RG. Soft tissue sarcoma. In DeVita VT, Hellman S, Rosenberg SA (eds). Cancer: Principles & Practice of Oncology. Philadelphia, Lippincott Williams & Wilkins, 2001, pp. 1841–1980.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012; 379: 1879–1886.
20. Chawla SP, Blay J, et al. Results of the phase III, placebo-controlled trial (SUCCEED) evaluating the mTOR inhibitor ridaforolimus (R) as maintenance therapy in advanced sarcoma patients (pts) following clinical benefit from prior standard cytotoxic chemotherapy (CT). Presented at the 2011 Annual Meeting of the American Society of Clinical Oncology.
21. Ciresa M, D’Angelillo RM, Ramella S, et al. Molecularly targeted therapy and radiotherapy in the management of localized gastrointestinal stromal tumor (GIST) of the rectum: a case report. Tumori. 2009; 95: 236–239.
22. Joensuu H. Predicting recurrence-free survival after surgery for GIST. Lancet Oncol. 2009; 10: 1025.
23. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006; 368: 1329–1338.
24. Bonvalot S, Eldweny H, Haddad V, et al: Extra-abdominal primary fibromatosis: aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol. 2008; 34: 462–468.
25. Fiore M, Rimareix F, Mariani L, et al: Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol. 2009; 16: 2587–2593.
26. Nuyttens JJ, Rust PF, Thomas CR Jr, et al. Surgery versus radiation therapy for patients with aggressive fibromatosis or desmoid tumors: a comparative review of 22 articles. Cancer. 2000; 88: 1517–1523.
27. Schlemmer M. Desmoid tumors and deep fibromatoses. Hematol Oncol Clin North Am. 2005; 19: 565–571.
28. Gebhardt MC, Hornicek FJ. Osteosarcoma and variants. In Bell MJ, et al (eds). Orthopaedics and Trauma. Oxford University Press, Oxford, 2002, pp. 224–238.
29. Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res. 1980; 153: 106–120.
30. DeLaney TF, Park L, Goldberg SI, et al. Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys. 2005; 61: 492–498.
31. Ciernik IF, Niemierko A, Harmon DC, et al. Proton-based radiotherapy for unresectable or incompletely resected osteosarcoma. Cancer. 2011; 117: 4522–4530.
32. Harsh GR, Janecka IP, Mankin HJ, et al. Chordomas and Chondrosarcomas of the Skull Base and Spine. Thieme Medical Publishers, New York, 2003.
33. Bjornsson J, McLeod RA, Unni KK, et al. Primary chondrosarcoma of long bones and limb girdles. Cancer. 1998; 83: 2105–2119.
34. Lee FY, Mankin HJ, Fondren G, et al. Chondrosarcoma of bone: an assessment of outcome. J Bone Joint Surg Am. 1999; 81: 326–338.
35. Goda JS, Ferguson PC, O’Sullivan B, et al. High-risk extracranial chondrosarcoma: long-term results of surgery and radiation therapy. Cancer. 2011; 117: 2513–2519.
36. Rosenberg AE, Nielsen GP, Keel SB, et al. Chondrosarcoma of the base of the skull: a clinicopathologic study of 200 cases with emphasis on its distinction from chordoma. Am J Surg Pathol. 1999; 23: 1370.
37. DeLaney TF, Liebsch NJ, Spiro IJ, et al. Proton radiotherapy for spine and paraspinal sarcomas. Int J Rad Oncol Biol Phys. 2006; 66: 115.
38. Schwab JH, Healey JH, Rose P, et al. The surgical management of sacral chordomas. Spine. 2009; 34: 2700–2704.
39. Keisch ME, Garcia DM, Shibuya RB. Retrospective long-term follow-up analysis in 21 patients with chordomas of various sites treated at a single institution. J Neurosurg. 1991; 75: 374–377.