Roland Haubner • Olaf Prante
INTRODUCTION
Over the past several decades, there has been a growing interest in radiopharmaceuticals to image molecular processes in oncology. Because the clinical success of 2-[18F]fluoro-2-deoxyglucose [FDG] to image glucose metabolism in tumors, an increasing number of tracers have been developed for potential clinical use. These radiotracers are designed to noninvasively provide information on a variety of processes including amino acid transport and protein synthesis, hypoxia, lipid metabolism, apoptosis, proliferation, and angiogenesis. Moreover, the determination of receptor overexpression especially of hormone receptors like somatostatin, bombesin, and cholekocystokinin receptors offers a sensitive diagnostic tool. Targeting these structures with radiolabeled peptides also provides an important treatment options, peptide receptor radionuclide therapy (PRRT). This chapter will briefly review the molecular processes leading to the accumulation of the corresponding radiopharmaceuticals and provide an overview of the most important radiolabeled compounds developed (Table 42.1).
GLUCOSE METABOLISM: IMAGING
Accumulation Mechanism and Synthesis of [18F]FDG
In the field of molecular imaging, positron emission tomography (PET) has emerged as the driving force of molecular imaging, significantly improving the treatment management of cancer patients including radiation treatment planning.71 The best known and most commonly used PET radiopharmaceutical is FDG, which assesses the metabolic state of malignant lesions. In FDG, the hydroxyl group in the 2-position of glucose is replaced by fluorine-18, providing a deoxyglucose analog with beneficial biochemical properties for imaging purposes. The deoxy carbohydrate analog [18F]FDG linearly accumulates in cells and the uptake is enhanced when increased glycolysis occurs. This is often associated with the increased growth rate and malignant potential of tumors.72 [18F]FDG is transported into the cell by one of several glucose transporters (in tumor cells, this is usually GLUT-1 or GLUT-3) and subsequently phosphorylated by hexokinase to give the 6-phosphate derivative. Because the hydroxyl group in position 2 of the molecule is missing, the subsequent reaction with the endogenous glucose-6-phosphate isomerase does not occur or is negligible. The negative charge of [18F]FDG-6-phosphate prevents the migration across the cell membrane (Fig. 42.1).
Glucose transport and metabolism by tumor cells is altered leading to increased [18F]FDG uptake compared to normal cells. One mechanism that facilitates the high glycolytic rate in tumor cells appears to be the overexpression of the glucose transporters.73 Increased activity of one or both subtypes of the mitochondrial enzyme, hexokinase, also contributes in tumor but not normal cells.74–76 However, malignant cells are not the only cells that show increased uptake of [18F]FDG.75,77,433 The significant uptake of glucose into the brain is a consequence of the high expression of GLUT-1 within the blood–brain barrier [BBB] enabling the high background glucose metabolism of normal grey matter structures and low tumor-to-background ratios when brain tumors are imaged with [18F]FDG PET. Due to repair processes in tissue after therapeutic interventions, wound healing, or alternative angiogenesis-related processes, such as arthritis, inflammation, or vascular diseases75,78,79 macrophages infiltrate the tumor, resulting in significant [18F]FDG uptake comparable to that of viable tumor cells.80,81 Up to 25% of the total [18F]FDG uptake measured in the tumor tissue is because of uptake in macrophages and granulation tissue.81 Using well-defined in vitro models, the mechanisms of [18F]FDG uptake have been studied in detail, improving our knowledge on various interesting parameters that trigger [18F]FDG uptake mainly by hypoxia-inducible transduction pathways.82,83 Hypoxia-inducible factor (HIF-α) increases uptake of [18F]FDG through activation of the glycolytic pathway. This notion was initially reported for hypoxic breast cancer cells.84 In summary, the analysis and interpretation of [18F]FDG PET data must be done with caution, considering the lack of specificity of [18F]FDG.75,80,81,85–87
The radiosynthesis of [18F]FDG was initially described by Ido et al.88 in 1977. Two different strategies have been described: The electrophilic fluorination of 3,4,6-triacetyl-D-glucal (TAG) using acetylhypofluorite89 and the aminopolyether-supported nucleophilic substitution of 1,3,4,6-tetra-O-acetyl-2-O-triflyl-β-D-mannose by Hamacher et al.1 At the present time, the latter strategy is most frequently employed because of the commercial availability of the [18F]FDG precursor, the high radiochemical purity and especially higher specific activity available, and the advantage of getting an epimeric pure product.90 There are various automated synthesis modules commercially available. These instruments provide high amounts of [18F]FDG in decay-uncorrected yields of approximately 70% within the relatively short overall reaction time of approximately 30 minutes.
AMINO ACID TRANSPORT AND PROTEIN SYNTHESIS: IMAGING
Imaging of brain tumors by PET has been realized using radiolabeled amino acids. Although lesion detection remains an important aspect, there is a growing interest in multimodal imaging, therapy planning, evaluating response to treatment, and providing prognostic information. The radiosyntheses of 11C- and 18F-labeled amino acids and their derivatives, including artificial amino acids, have been extensively described. These studies include the naturally occurring L-[11C]leucine,91 L-[methyl-11C]methionine ([11C]MET),92 and L-[1-11C]tyrosine93 as well as unnatural aliphatic (e.g., [11C]AIB) and alicyclic (e.g., [11C]ACPC) amino acids (for review, see McConathy and Goodman94). The most commonly used 11C-labeled amino acid is [11C]MET. The radiosynthesis of [11C]MET can be achieved by various synthetic pathways that rely on the S-alkylation of the corresponding sulfide anion, using either [11C]methyl iodide or [11C]methyl triflate.2 Because variations in the radiosynthesis do not significantly affect the specific activity or the enantiomeric, radiochemical, and chemical purity, these procedures appear to be very robust indeed.
TABLE 42.1
MOST IMPORTANT COMPOUNDS FOR TUMOR IMAGING AND THERAPY USED IN NUCLEAR MEDICINE


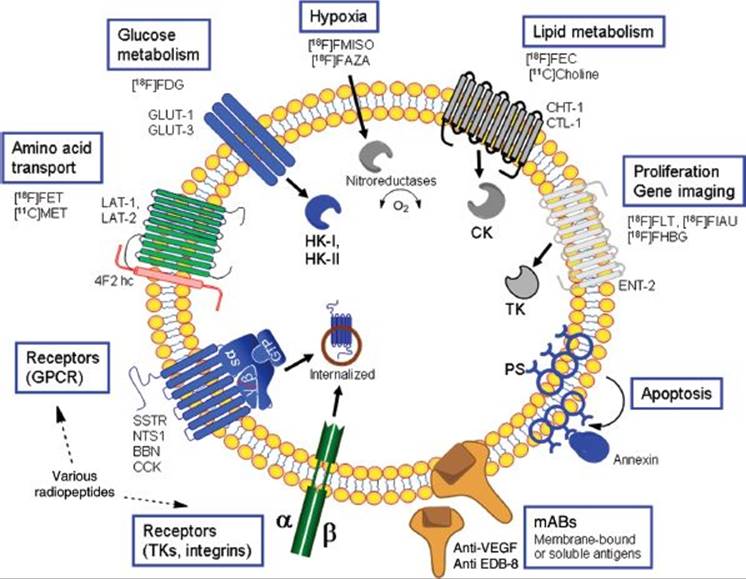
FIGURE 42.1. Overview of tracer concepts for diagnosis and therapy in oncology. The main pathways are stated in the blue frames and related classical radiopharmaceuticals are given; other important radiopharmaceuticals are provided in Table 42.1. Details about the different uptake mechanisms are found in the corresponding parts of the text. 4F2hc, CD98 (4F2 heavy chain); BBN, bombesin; CCK, cholecystokinin; CLT-1, choline transporter–like protein 1; CHT-1, high-affinity choline transporter; ED-B, extra domain-B; ENT-2, equilibrative sodium-independent nucleoside transporter-2; GLUT, glucose transporter; GPCR, G-protein–coupled receptors; HK, hexokinase; LAT, L-type amino acid transporter; NTS1, neurotensin receptor subtype 1; PS, phosphatidylserine; SSTR, somatostatin receptor; TK, thymidine kinase; TKs, tyrosine kinases; VEGF, vascular endothelial growth factor.
In contrast to the radiosyntheses of 11C-labeled amino acids, the development of 18F-labeled amino acids appeared to be more complicated. Today, O-(2-[18F]fluoroethyl)-L-tyrosine ([18F]FET) and 3,4-dihydroxy-6-[18F]fluoro-L-phenylalanine ([18F]FDOPA) are the main candidates for further clinical studies. [18F]FET can be produced either in a two-step synthesis directly from the disodium salt of L-tyrosine using 2-[18F]fluoroethyl tosylate as a labeling synthon or by direct nucleophilic 18F-for-tosylate substitution on a protected O-(2-tosyloxyethyl)-L-tyrosine derivative in uncorrected radiochemical yields of more than 20%.3,4 This method is of interest because it lends itself to commercial regional transport of the tracer and clinical utilization.95 [18F]FDOPA is most frequently produced via electrophilic radiofluorodestannylation of N-formyl-3,4-di-tert-butoxycarbonyl-6-(trimethylstannyl)-L-phenylalanine using [18F]F2 followed by acidic removal of the protecting groups.5
Uptake Characteristics of Radiolabeled Amino Acids
Initially, amino acid tracers were developed with the intention to measure protein synthesis rates,96 when the most interesting candidates were tyrosine analogs. However, the rate of amino acid transport rather than the protein synthesis rate seems to be the major determinant of tracer uptake in tumor imaging studies (Fig. 42.1). The expression of amino acid transporter subtypes on tumor cells appeared to be manifold. They differ with regard to substrate specificity and sodium dependency.97–99
To date, the development of PET imaging agents has mainly focused on substrates of systems L (leucine preferring) and/or A (alanine preferring).94 The transport system L is sodium-independent and recognizes amino acid substrates with large neutral side chains. There are four isotypes of transporters, called LAT1 to LAT4.100–103 LAT1 and LAT2 form heteromeric complexes with the heavy chain of 4F2 antigen (4F2hc, CD98), a single-transmembrane domain protein essential for the functional expression of LAT1 and LAT2. LAT1 and LAT2 are sodium-independent obligatory exchangers, where the substrate transport occurs in exchange for other amino acid substrates and the transporter activity depends on the presence of transporter substrates inside the cell. The LAT1 isotype prefers large neutral amino acids with aromatic side chains, whereas LAT2 exhibits broad substrate selectivity and mediates the transport of small neutral amino acids, such as serine. Systems A and ASC (alanine, serine, cysteine) are sodium-dependent and transport small neutral amino acids. However, radiolabeled amino acid analogs are usually transported into the cell by more than one transporter system as the different amino acid transporter subtypes demonstrate overlapping substrate specificity. This is also the case for [11C]MET, which is a substrate for system L and, to a lesser extent, for systems A and ASC. In contrast to system L, substrates with a bulky side chain are preferably transported via the system L. But in contrast to natural L-amino acids most fluorinated amino acids are not incorporated in proteins.
Cellular [18F]FET uptake is mainly attributed to transport via system L using the in vitro model of SW 707 colon carcinoma cells.86 In addition, Langen et al.104 described the sodium-dependent transport of [18F]FET in the rat glioma cell line F98 that could be ascribed mainly to system B0,+, besides sodium-independent transport via system L. Stöber et al.105 provided evidence that System L–mediated FET uptake could be beneficial for the differentiation of tumor and inflammation.
Nevertheless, in vivo PET imaging studies comparing [11C]MET and [18F]FET in brain tumor patients showed similar distribution patterns and similar tumor-to-blood ratios.95 An advantage of using unnatural instead of natural amino acids is the improved metabolic stability of the unnatural amino acid thus avoiding problems with metabolites which complicate the kinetic analysis of PET imaging data. Moreover, animal studies suggest that [18F]FET, in contrast to [18F]FDG and [11C]MET, is not accumulated in inflammatory tissue, making it potentially superior for distinguishing neoplasm from inflammation.103,106 Moreover, the first study to investigate the use of FET PET in peripheral tumors reported better distinction between squamous cell carcinomas and inflammatory tissues with FET PET than with FDG PET.107
FET PET has been successfully employed in the diagnostic workup of brain tumors.108–110 These studies used multimodal imaging, for example, MR imaging (also for spectroscopic analysis of brain markers) and correlation to FET PET.108,111,112 Differences in the uptake of [18F]FET and [11C]MET in inflammatory tissue are not understood. It is tempting to speculate that the difference could be caused by a different expression pattern of transporter system isotypes.95,113
[18F]FDOPA is an 18F-labeled analog of the endogenously occurring L-DOPA and has been extensively used to evaluate the dopaminergic system in the brain using PET. Besides the physiologic high uptake of [18F]FDOPA in the substantia nigra and striatum,114 [18F]FDOPA shows high uptake in primary brain tumors caused by increased amino acid transport. The transporter responsible for [18F]FDOPA uptake is reported to be the System L.115 As the enzyme aromatic amino acid decarboxylase (AADC) recognizes [18F]FDOPA as a substrate, it can be used also to image well-differentiated metastatic neuroendocrine tumors (NETs) that express AADC but lack other specific molecular markers.116,117
HYPOXIA: IMAGING
Hypoxia Imaging: Tracers
In radiotherapy planning, tumor-cell hypoxia has been understood to be one of the most important factors resulting in resistance to treatment. The hypoxic cell fraction appears to be an independent risk factor for tumor progression. In vivo imaging of the hypoxic cell fraction and oxygenation in tumors is of growing interest in planning radiation treatment.
Currently, the most commonly used radiotracers for hypoxia PET imaging are 1-[2-nitro-1-imidazolyl)-3-[18F]fluoro-2-propanol ([18F]FMISO), 1-(5-[18F]fluoro-5-deoxy-α-D-arabinofuranosyl)-2-nitroimidazole ([18F]FAZA) and [64Cu]copper(II)-diacetyl-bis (N4-methylthiosemicarbazone [64Cu]ATSM. The radiosyntheses of the nitroimidazole derivatives [18F]FMISO and [18F]FAZA are achieved by nucleophilic 18F-fluorination of 1-(2-nitro-1-imidazolyl)-2-O-tetrahydropyranyl-3-O-toluene-sulfonylpropane-diol and 1-α-D-(2,3-diacetyl-5-tosyloxy-arabinofuranosyl)-2-nitroimidazole, respectively. After the radiolabeling step, the protective groups are efficiently cleaved and the corresponding product is isolated by reverse-phase HPLC. Both tracers can be produced in high specific activity and radiochemical purity. The radiochemical yields, however, are much higher for [18F]FMISO (80%; decay-corrected) than for [18F]FAZA (20%; decay-corrected).6
The straightforward radiosynthesis of [64Cu]ATSM is performed by incubating the cyclotron product [64Cu]CuCl2 in a buffered solution in the presence of H2ATSM at room temperature.7 The product [64Cu]ATSM is obtained in high radiochemical purity after employing a solid-phase extraction procedure for isolation of the final product. Despite the limited availability of the radionuclide Cu-64, because of the limited number of high-energy cyclotrons that are capable of producing Cu-64, the radiosynthesis of the hypoxia tracer [64Cu]ATSM has some major advantages: (a) The incubation step with the radioisotope is much easier; (b) the labeling step is much faster compared to the syntheses of [18F]FMISO and [18F]FAZA; and (c) complicated and laborious deprotection and purification steps are not needed. One disadvantage of Cu-64 might be its rather long half-life of 12.7 hours, which results in higher radiation dose to healthy organs of the patient. Because the investigational new drug application for [64Cu]ATSM was recently approved by the FDA, there is increasing interest in the outcome of a multicenter trial to validate this tracer which could lead to clinical approval.
Hypoxia Imaging Tracers: Biologic Characteristics
[18F]FMISO has a partition coefficient near unity indicating that this molecule is able to penetrate almost all cell membranes by diffusion (Fig. 42.1). After cellular uptake, intracellular nitroreductases transfer an electron to the nitro group of the imidazole. In normoxic cells, the reductive reaction is hindered by the presence of oxygen that acts as an electron scavenger and [18F]FMISO thus maintains its original structure allowing free diffusion. Under hypoxic conditions, a second electron transfer reduces the nitroimidazole to a highly reactive intermediate, capable of binding to proteins and RNA. Thus [18F]FMISO gets metabolically trapped inside the cell under hypoxic conditions. Consequently, [18F]FMISO uptake is inversely related to the partial pressure of intracellular oxygen.118 This is also true for the accumulation of [18F]FAZA by hypoxic cells. Because of the glycosyl moiety in the chemical structure of [18F]FAZA, this tracer is more hydrophilic than [18F]FMISO. This is also reflected by an increased renal clearance of [18F]FAZA as compared to the lipophilic tracer [18F]FMISO which in turn suffers from slow uptake kinetics, low signal-to-noise ratios, and thus the need for imaging studies at late time points after tracer injection (up to 4 hours p.i.). The hydrophilic tracer [18F]FAZA results in a higher contrast between hypoxic and normoxic tissues.119However, the quality of [18F]FAZA PET images from patient studies do not appear to be superior. Thus, the earlier enthusiasm for [18F]FMISO has not been replaced by [18F]FAZA so far. Further studies that focus on the quantitative image analysis may demonstrate which compound is superior and whether alternative PET tracers derived from nitroimidazole could provide even more advantages for hypoxia PET imaging.
In the case of [64Cu]ATSM there are several putative trapping mechanism.120 More recently, a slightly revised trapping mechanism has been suggested based on chemical, electrochemical, spectroscopic, and computational concepts.121 Presumably, [64Cu]ATSM enters the cell by passive diffusion independent of the oxygenation status of the cell. Intracellularly, [Cu(I)ATSM]− is generated in normoxic as well as hypoxic cells by enzyme-mediated reduction. Under normoxic conditions, rapid and facile reoxidation to the neutral Cu(II) complex [64Cu]ATSM occurs, which can leave the cell leading to an equilibrium between outer and inner concentrations of the tracer. In hypoxic cells, depending on the pH, unstable [Cu(I)-ATSMH] is formed. Then, [Cu(I)-ATSMH] rapidly dissociates allowing the interaction of Cu(I) with proteins inside the cell leading to the final trapping products. The tracer properties and the capacities of [18F]FMISO and [64Cu]ATSM for monitoring the oxygenation status in the tumor have been comparatively studied with murine tumor models and, more recently, via electrochemical modeling.122,123
It has been shown in squamous cell carcinoma (SCCVII)-bearing mice, that [64Cu]ATSM uptake was unable to predict changes in hypoxia when the oxygenation status was modulated, whereas the [18F]FMISO tumor uptake was more responsive to changes of the oxygenation status.124 A comparative study on [18F]FMISO and [64Cu]ATSM uptake in rats bearing an anaplastic prostate tumor (R3327-AT) revealed a poor correlation of the tracer’s intratumoral distribution when [18F]FMISO uptake at 4 hours post injection (p.i.) was compared with [64Cu]ATSM uptake at 2 hours p.i. but a good correlation when [18F]FMISO uptake 4 hours p.i. was compared with [64Cu]ATSM uptake at 16 hours p.i.125 This observation has been explained by the significant temporal evolution of [64Cu]ATSM uptake between 30 minutes and 20 hours after injection in this tumor type. Interestingly, in rats bearing a human squamous cell carcinoma (FaDu) the observed difference was negligible, when the early and the late [64Cu]ATSM micro-PET images were comparable and in accordance with the [18F]FMISO images. These observations suggest that the accumulation mechanism of both tracers differ and that further studies are needed for a final assessment. Recently, Bowen et al.123 used computational modeling to characterize the differences between the uptake of [64Cu]ATSM and [18F]FMISO in hypoxic tissue. Their results show that the high dynamic range of [64Cu]ATSM uptake was representative of a narrow range of low oxygen tension (highly dependent on the pH), whereas [18F]FMISO uptake was representative of a wide range of pO2 values that were independent of acidity.
LIPID METABOLISM: IMAGING
Substrates of Choline Kinase as Imaging Probes
A variety of choline derivatives are specific substrates of choline kinase, which is an enzyme that is overexpressed in malignant lesions. The phosphorylation reaction mediated by choline kinase leads to intracellular trapping of the respective substrates of the kinase, which in turn represents a suitable trapping mechanism for imaging probes. Choline was initially labeled with 11C-carbon, producing an isotopic radiotracer of the endogeneous substrate. The radiosynthesis of [11C]choline is achieved by two different strategies.8 Both reaction pathways rely on the use of 2-(dimethylamino)-ethanol (DMAE) which is 11C-methylated by either [11C]methyliodide or [11C]methyltriflate. These reactions are carried out either in solution, in the void volume of a solid-phase cartridge or by loading DMAE into a loop system. Following these strategies, radiochemical yields up to 95% are obtained. [11C]choline is routinely used in PET centers with nearby cyclotron access. A variety of tumors including, most prominently, prostate cancer, have been imaged.126 18F-labeled derivatives of choline have been synthesized to take advantage of the longer half-life of 18F. These derivatives include the analog tracers [18F]fluoromethylcholine ([18F]FCH) and [18F]fluoroethylcholine ([18F]FECH). In contrast to the radiosynthesis of [11C]choline, the tracers [18F]FECH and [18F]FCH are obtained by a two-step process. In the case of [18F]FECH,10 first, 2-[18F]fluoroethyl tosylate is synthesized by nucleophilic 18F-fluorination of 1,2-bis(toxyloxy)ethane; second, the N-alkylation of DMAE with 2-[18F]fluoroethyl tosylate is performed by using DMAE as the solvent of this reaction, to give [18F]FECH with a decay-corrected radiochemical yield of this synthetic strategy of about 46%. The synthesis of [18F]FCH begins with [18F]bromofluoromethane, which is produced via nucleophilic 18F-substitution of dibromomethane,9 and then further reacted with DMAE. Alternatively, [18F]bromofluoromethane can be quantitatively converted to [18F]fluoromethyl triflate in an online capillary system and then used for the final alkylation of DMAE, resulting in radiochemical yields of about 30% to 40%.
As an alternative tracer for prostate cancer imaging, radiolabeled acetate has been proposed. Similar to choline, the compound has been labeled either with 11C resulting in [11C]acetate ([11C]ACE) or with 18F, providing the analog tracer [18F]fluoroacetate ([18F]FAC). The radiosynthesis of [11C]ACE is reported as a Grignard reaction, starting with [11C]CO2 that is bubbled through a methyl magnesium bromide solution.11 After hydrolysis under acidic conditions, the desired product is isolated in radiochemical yields up to 70%.127 The radiosynthesis of [18F]FAC, however, is performed by the nucleophilic 18F-fluorination on ethyl O-mesylglycolate, when the 18F-product is subsequently hydrolyzed with sodium hydroxide to provide the final product in radiochemical yields of about 55%.11
Tracers for Choline Kinase: Biologic Characteristics
The overexpression of phosphorylcholine has been reported for a variety of malignant tumors, whereas only low levels are found in normal tissue.128 The presence of phosphorylcholine occurs first during the incorporation of choline into phospholipids by the Kennedy pathway.129 However, it is unclear if the choline kinase reaction or an upstream transporter significantly determines the tracer accumulation (Fig. 42.1). The choline transporter recognizes structural analogs of endogeneous choline as well as fluorinated choline derivatives. Two methyl groups are essential for substrate recognition by the enzyme; however, the third methyl group can be replaced by elongated alkyl chains.130Similar results have been obtained by studying the substrate flexibility of choline kinase.131 In addition to the two methyl groups, the hydroxyethyl group is important but the third methyl group can be replaced by alkyl chains. These data indicate that uptake mechanisms for [11C]choline, [18F]FCH, and [18F]FECH are rather similar.72 However, comparative in vitro tracer uptake studies with PC-3 prostate tumor cells show higher uptake of [11C]choline and [18F]FCH compared to [18F]FECH.9 In the clinical setting, tracer uptake usually reaches a plateau within 10 to 20 minutes after tracer injection, indicating that the transport of the tracer may be the rate-determining step, which is confirmed by other studies suggesting that choline transport and not the subsequent phosphorylation is the key step that determines choline uptake in cancer cells.72
It is well known that fatty acids are the primary metabolic energy source in the myocardium. 11C-labeled acetate ([11C]ACE) is rapidly metabolized to [11C]CO2 via the tricarboxylic acid cycle and released from cells. However, [11C]ACE has been shown to steadily accumulate in prostate cancer and other tumors. Thus, it is assumed that [11C]ACE enters lipid synthesis in cancer cells and is trapped intracellularly rather than proceed to [11C]CO2. Besides [11C]ACE, the use of fluoroacetate has been proposed. However, fluoroacetate is a toxic compound originally found in the South African poison plant Dichapetalum chymosum. Similar to acetate, fluoroacetate is a substrate for acetyl coenzyme A synthetase but with lower specificity.132 Its toxicity is because of its conversion to fluorocitric acid, which is an inhibitor of the tricarboxylic acid cycle. Ponde et al. calculated that maximum [18F]FAC concentration in patients is 10−6 times the human LD50. In animal studies, uptake in prostate cancer is very similar to [11C]ACE.11 However, because of its lack of oxidative metabolism [18F]FAC shows slower clearance than [11C]ACE into the intestine and the urinary bladder and is only rarely used in clinical settings. The radiation dose associated with [18F]FAC/PET imaging is well within the accepted limits,133,134 which might facilitate clinical utilization of [18F]FAC in the future.
IMAGING PROLIFERATION
Tracers for Imaging Proliferation
The proliferation rate of tumor cells can be studied with [11C]thymidine and 3′-[18F]fluoro-3′-deoxythymidine ([18F]FLT), a thymidine deoxy analog where the hydroxyl function in position 3′ is replaced by 18F-fluorine.135 However, the rapid metabolism of [11C]thymidine and the need for complicated image acquisition and analysis with correction of the blood input function is an obstacle to the general acceptance of [11C]thymidine as a proliferation marker in PET imaging studies in routine clinical practice.136,137 As a practical alternative, [18F]FLT and other analogs have been developed.
Several approaches to the radiosynthesis of [18F]FLT have been developed. These strategies differ in the choice of precursor, but the activation at position 3′ in the molecule is the prerequisite for the nucleophilic 18F-substitution reaction. There are promising strategies that suggest 2,3′-anhydrothymidine derivatives or 3′-O-nosylthymidine derivatives as the precursor of choice. Because hydroxyl groups are thought to decrease the nucleophilicity of the [18F]fluoride, all the reported synthesis routes so far use 5′-benzoyl or dimethoxytrityl derivatives. Using 3-N-dimethoxybenzyl-5′-dimethoxytrityl-3′-O-nosylthymidine as a precursor, Grierson and Shields138 described a three-step synthesis of [18F]FLT, consisting of the nucleophilic substitution of the nosyl leaving group, removal of the protection groups by ceric ammonium nitrate, and isolation of the final product by RP-HPLC, providing the final [18F]FLT within 90 minutes in about 8% yield. Alternatively, Machulla et al.12 used the precursor 5′-O-(4,4′ dimethoxytrityl)-2,3′-anhydrothymidine for the reliable production of [18F]FLT within 90 minutes in about 7% yield. A further improvement has been described by the use of the precursor 3-N-Boc-1-[5-O-(4,4′-dimethoxytrityl)-3-O-nosyl-2-deoxy-β-D-lyxofuranosyl]thymidine,139 applying a three-step procedure with the labeling step, deprotection, neutralization with sodium hydroxide, and HPLC purification. This course of the synthesis of [18F]FLT provided the final product within 60 minutes with a 21 ± 4% yield.
Imaging Proliferation Probes: Biologic Characteristics
Similar to thymidine, [18F]FLT enters the cell via the nucleoside transporter and to a lesser extent via passive diffusion,137 where the rate-limiting step of [18F]FLT uptake is the initial phosphorylation by thymidine kinase-1 (TK-1) (Fig. 42.1).135 In principle, further phosphorylation is possible, but because of the missing 3′-hydroxyl group, only negligible amounts of the tracer are incorporated into DNA. The negative charge of the phosphate group prevents the tracer from penetrating biologic membranes and thus, phosphorylated [18F]FLT is trapped inside the cell. Besides the uptake mechanism, there is some dephosphorylation via 5′-deoxynucleotidases, but this pathway can be neglected because it is relatively slow when compared to the thymidine kinase activity. Therefore, the uptake of [18F]FLT reflects TK-1 activity, which is the parameter that is addressed and analyzed by [18F]FLT-PET imaging studies.140
Concerning the biologic environment, it is well known that thymidine kinase activity differs during the cell cycle, that is during the S-phase high levels of thymidine kinase are observed and the number of proliferating and malignant cells reveal an increased enzyme expression upto 15-fold compared to normal cells.137 As uncontrolled proliferation is a biomarker of cancer cells, there is great interest in imaging tumor-cell proliferation, especially in the context of therapeutic planning and intervention.135,141 However, only a small fraction of cells in a variety of solid tumors demonstrate the S-phase in their proliferation status. Consequently, the accumulation of [18F] FLT has been found to be markedly lower than that of [18F]FDG in most solid tumors. This makes [18F]FLT less suitable for tumor staging by PET imaging. However, it is expected that [18F]FLT may play an important role in monitoring tumor response to treatment.141 On the one hand, preclinical studies have repeatedly indicated that arrest of tumor cells in the G1-phase causes a rapid and significant decrease in [18F]FLT uptake. For example, changes in tumor proliferation assessed by [18F]FLT-PET early after initiating docetaxel chemotherapy in breast cancer has been recently shown to predict lesion response midtherapy with good sensitivity.142 On the other hand, putative active agents for the treatment of cancer could cause a temporary increase in [18F]FLT uptake known as a flare response. For example, chemotherapeutic agents that block thymidine synthesis have been shown to cause activation of thymidine kinase activity and [18F]FLT uptake.143 Further studies that compare [18F]FLT with [18F]FDG PET, including multicenter clinical trials, will be important to evaluate [18F]FLT as an imaging probe to predict tumor response to therapy.141,144
Gene Imaging Via Nucleoside Derivatives
The mechanism of phosphorylation of nucleosides is also proposed to be used to noninvasively monitor gene expression by reporter gene imaging techniques. Such techniques would allow localization as well as determination of the duration and magnitude of gene expression during gene therapeutic approaches. Other strategies use these techniques to monitor properties of cell trafficking including metastasis, stem cell transplantation, and lymphocyte response to inflammation in a variety of preclinical settings145 but also in initial clinical studies.146 Noninvasive reporter gene imaging involves a reporter transgene placed under the control of upstream promoter/enhancer elements. These promoter/enhancer elements can always be “turned on” with constitutive promoters, or can be sensitive to activation by specific endogenous transcription factors.147
Several attempts with different reporter gene constructs, including the sodium iodine symporter gene,148 the somatostatin receptor (SSTR) gene,149 and the norepinephrine transporter gene,150 have been studied. The most extensively studied approach uses the herpes simplex virus type 1-thymidine kinase (HSV1-tk) gene as a reporter gene and different radiolabeled endogenous nucleosides as reporter probe.145,151 The reporter probe is designed to be exclusively recognized by the HSV1-tk and not by human thymidine kinase, as [18F]FLT and other proliferation imaging agents do, resulting in a selective accumulation of the reporter probe only where the reporter gene is expressed. Because of the design of the reporter gene construct, the reporter gene expression correlates with the expression of the gene of interest.
There are two different classes of radiolabeled reporter probes to image HSV1-tk expression: (a) Pyrimidine nucleoside analogs (e.g., FIAU152 and FEAU153) as well as (b) acycloguanosine derivatives (e.g., FPCV,154 FHBG,155and FHPG156).
With the exception of FIAU all the compounds are labeled with 18F-flourine. FIAU also has been labeled with radioiodine isotopes. The advantage of this labeling approach is that the synthesis of the tracer is much easier. Radiolabeling can be carried out by direct iodination of FAU (2′-fluoro-2′-deoxy-1-β-D-arabinofuranosyluracil)157 or FTAU (5-trimethylstannyl-1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl) uracil).158 The first was labeled using the Iodogen method at 70°C within 10 minutes.152 Subsequently, the mixture is passed through a C18-cartridge and final separation from the precursor was carried out using HPLC. To label the second compound, acetic acid/hydrogen peroxide is used for oxidation in a sodium hydroxide/chloroform solution with 30-second ultrasound treatment at room temperature.13 Isolation of the product is again carried out by HPLC. Altogether, both techniques are straightforward. However, the tin-precursor method allows preparation of the tracer in much higher radiochemical yield compared to the Iodogen method (∼70% versus 15%).
Because of the need for protection groups, labeling of the 18F-derivatives is always a multistep process with a final deprotection step.159 Synthesis of the most prominent derivatives, FHBG and FHPG, starts with labeling a precursor like N-(p-anisyldiphenylmethyl)-9-[(4-(p-toluolsulfonyloxy))-3-panisyldiphenylmethoxy-methylbutyl] guanine for FHBG and N-(p-anisyldiphenylmethyl)-9-[[1-(p-anisyldiphenylmethoxy)-3-(p-toluolsulfonyl-oxy)-2-propoxy]methyl] guanine for FHPG. The main steps of the synthesis of both compounds are very similar. In general, after azeotropic distillation to get water-free 18F-fluorine, the precursor in acetonitrile is added and the reaction mixture is heated to temperatures between 100° and 140°C using reaction times between 10 and 30 minutes. For deprotection, 1 N acetic acid or hydrochloric acid at temperatures of approximately 120°C is used. After neutralization with sodium hydroxide the product is finally isolated preferably using HPLC systems. The uncorrected radiochemical yields are comparable to the FIAU synthesis using the Iodogen method and range between 10% and 20%.
There is an open discussion about which compound is most suitable to image HSV-tk expression. One in vitro and in vivo study demonstrated, that the radioiodinated uracil nucleoside [*I]FIAU has a significantly higher specific accumulation than the acycloguanosine derivative [18F]FHPG.13 Similar results were found by Tjuvajev et al.,159 comparing FIAU, FHPG, and FHBG. Again, in vitro and in vivo results including small-animal PET imaging showed that FIAU is the more efficient reporter probe compared to the acycloguanosine derivatives. This suggests that [124I]FIAU seems to be the preferred probe for PET imaging of HSV1-tk gene expression. One approach that improves imaging with aycloguanosine derivatives is based on the introduction of a mutant HSV1-tk enzyme.14 The used HSV1-sr39tk enhanced the [18F]FHBG uptake twofold compared with the wild type HSV1-tk.
The synthesis of 18F-labeled FIAU has been described. In contrast to the 18F-labeling strategies for acycloguanosine derivatives, the synthesis of [18F]FIAU starts with the labeling of the sugar moiety which is subsequently conjugated with the uracil derivative, making this a very complex synthesis, inserting the radiolabel in the initial synthesis step. Most recently, an improved synthesis for a pyrimidine-based tracer carrying out the multistep synthesis using a one-pot strategy is described.160 This approach leads to the desired products with yields ranging between 4% and 10% (decay-corrected) and reduces reaction time from 3.5 to 2.5 hours.
APOPTOSIS: IMAGING
Biologic Characteristics
Apoptosis (programmed cell death) is a well-studied tumor response to radiation and/or chemotherapy. Thus there is a keen interest in imaging strategies to allow noninvasive monitoring of the apoptotic pathways. A variety of molecular processes are involved in these pathways.161 A family of aspartate-specific cysteine proteases, caspases, are another target structure for tracer development. Caspases are involved in the central pathway of apoptosis.162Two different activation pathways are involved: The intrinsic, or mitochondrial and the extrinsic pathway. The central caspase in both pathways is caspase 3, which activates poly-ADP-ribose polymerase and slices DNA.
Another target structure involved in changes in the normal plasma membrane asymmetry during apoptosis is the activation of caspase 3 which induces a rapid redistribution of phospholipids.161,163 In vital cells, the two enzymes, flippase and floppase, that control anionic phospholipids like phosphatidylserine (PS) are restricted to the inner choline phospholipids and to the outer membrane leaflet (Fig. 42.1). In apoptotic cells, flippase and floppase are shut off and a third enzyme known as scramblase is activated. This enzyme allows redistribution of phospholipids across the lipid bilayer resulting in the presentation of high amounts of PS to the extracellular space.
Available Imaging Probes
Based on insights into the apoptotic process the most intensively studied tracers are annexin V derivatives. Annexin V is a naturally occurring 36-kDa human protein (319 AA) that binds with high affinity to externalized PS on apoptotic cells.164 Most clinical studies are focused on the development of SPECT tracers labeled with 99mTc.165 For initial human trials, the penthioate radioligand method (N2S2) was used to label annexin V.15 This method starts with the formation of 99mTc-gluconate to synthesize a 99mTc-N2S2-TFP ester. The activated ester randomly reacts with amino functions of lysine in the protein sequence leading to 99mTc-labeled annexin V. Because of the complexity of this synthesis route, improved labeling strategies have been developed. One approach introduced the bifunctional hydrazine nicotinamide (HYNIC).16 The HYNIC-Annexin V is produced by conjugation of the succinimidyl 6-hydrazinopyridine-3-carboxylate hydrochloride with amino functions of the protein (preferable from lysine side chains).166 The labeling was carried out by incubating the protein with 99mTc-pertechnetate in a Sn/tricine solution. This results in labeled HYNIC-Annexin V in yields greater than 93% and a specific activity of 7.4-MBq/μg protein. Similar to the N2S2-Annexin derivative, 99mTc-HYNIC-Annexin V showed greatest uptake in kidneys, liver, and urinary bladder but demonstrated no biliary excretion.167 Reduction in renal uptake can be achieved by introduction of mutant forms. These mutants (V-117 and V-128) have an endogenous site for 99mTc binding consisting of six amino acids added at the N-terminal end of the protein.168 A marked increased in vivo localization to apoptotic sites was found for these mutants.169
Alternative strategies use 111In to label annexin V. Annexin is not only conjugated with DTPA for chelation but also with polyethylene moieties improving the blood half-life of the compound.17 However, high nonspecific uptake which made further studies necessary.
As indicated, most studies have focused on tracers for SPECT imaging but there have been some efforts to develop PET tracers. N-succinimidyl 4-fluorobenzoate has been used to produce 18F-annexin V.18 This tracer showed lower activity accumulation in liver, spleen, and kidneys compared to 99mTc-HYNIC annexin V. Another approach used site-specific derivatization to allow labeling via 18F-maleimide to produce mutant 18F-annexin V-117 and 18F-annexin V-128.19 Most recently, the same approach was also used for 68Ga labeling. Using site-specific mutagenesis, Cys-2-Anx A5 and Cys-165-Anx A5 containing a single available cysteine residue at either position 2 or position 165 was produced, which was labeled by using 68Ga-DOTA-maleimide as prosthetic group.170 However, further preclinical and subsequent human studies are needed to demonstrate their potential in imaging apoptosis in routine clinical settings.
Another target structure used for tracer development is caspase-3. The studies are mainly focused on isatin class caspase-3 inhibitors, which bind via the dicarbonyl function of the isatin moiety to the cysteine residue of the caspase-3 active site.171 A variety of potential labeling precursors have been synthesized which produced [18F]fluoroethyl phenyl ether derivatives as a putative PET tracer.172,173 Further efforts led to 1-[18F]fluoro-2-hydroxyethyl benzyl derivatives.174 However, synthesis yields and specific activity are low; thus further studies are needed before these tracers can be used for clinical apoptosis imaging. [18F]ICMT-11 is another compound selected from an isatin-5 sulfonamide library. It exhibited high caspase-3 affinity and high metabolic stability.175 The radiolabeling is straightforward using click chemistry. 2-[18F]Fluoroethylazide was used for the copper-catalyzed cycloaddition and conjugated with the corresponding alkyne precursor. In murine tumor models, drug treatment–increased tracer uptake indicating that this class has potential to image caspase-3–induced apoptosis. Studies have been limited to several animal models and no published data from patients are available. The in vivo specificity of this class of tracers is unclear.
ANGIOGENESIS: IMAGING
Angiogenesis is a process involving the degradation, growth, and spreading of new blood vessels. It occurs during embryogenesis, female reproductive cycle, tissue remodeling, and wound healing and plays a pivotal role in nontumor-associated processes of growth and development. A variety of diseases are characterized by an imbalance of the angiogenic process176,177 including psoriasis,178 restenosis,179 rheumatoid arthritis,180 diabetic retinopathy,181and tumor growth.182 Angiogenesis research is one of the most rapidly growing biomedical disciplines and strategies to alter angiogenesis have already been evaluated in clinical trials. It is estimated that more than 500 million patients potentially benefit from such strategies.183 These developments are prime examples of targeted therapy. Imaging has a potential role to determine the role of angiogenesis as a predictive component in clinical medicine.184
The so-called angiogenic switch is regularly triggered by an insufficient oxygen supply resulting in cell hypoxia and activation of the hypoxia-inducible factor (HIF-α) and hypoxia response elements that in turn activate the expression of vascular endothelial growth factor (VEGF), a key component of angiogenesis.185 There are many additional regulating factors such as fibroblast growth factor (bFGF, aFGF) and platelet-derived endothelial cell growth factor (PDGF) that are secreted by a variety of cells.177,186 After activation of the endothelial cells, proteolytic enzymes such as matrix metalloproteinases (MMPs) are produced to degrade the basement membrane and the extracellular matrix (ECM), providing sufficient space for the sprouting vessels.187,188
Most important for imaging purposes are the integrins.189–191 These receptors are involved in endothelial cell migration, adhesion and growth, survival, and differentiation. It has been demonstrated that antibodies as well as low–molecular-weight antagonists, recognizing the integrins αvβ3 and αvβ5, block angiogenesis in murine tumor models and induce apoptosis not only of activated endothelial cells, but also of αvβ3-positive tumor cells.192–195 However, based on several knockout experiments, there is evidence that αvβ3 and αvβ5 are antiangiogenic or negative regulators of angiogenesis rather than proangiogenic.196
However, currently available imaging techniques are limited to monitoring the therapeutic success of antiangiogenic drugs as novel therapeutics for the treatment of tumors.197–199 At present, most of the work on the development of PET tracers to image angiogenesis is focused on radiolabeled αvβ3-antagonists, MMP inhibitors, antibodies targeting VEGF, and on single-chain Fv antibody fragments selectively binding to a fibronectin isoform.
Imaging Integrin Expression: RGD Peptides
The family of integrins includes 24 different heterodimeric receptors, derived from 18α- and 8β-mammalian subunits. Among these, the ανβ3-integrin is expressed on the surface of migrating cells,192 and plays an essential role in the regulation of tumor growth, local invasiveness, and metastatic potential.200,201
ECM proteins such as vitronectin, laminin, and fibronectin interact via the amino acid sequence Arg-Gly-Asp (RGD) with the integrin.202 Based on these findings, Aumailley et al.203 developed the αvβ3-targeting pentapeptide cyclo(-Arg-Gly-Asp-DPhe-Val-) which is the most prominent lead structure for the development of radiotracers targeting the ανβ3-integrin.204
MONOMERIC TRACERS: RADIOCHEMICAL STRATEGIES
In general, the principal approach to label peptides with 18F-fluorine relies on the use of prosthetic groups. The most prominent 18F-labeled tracer to image αvβ3 expression is [18F]Galacto-RGD, which is labeled via conjugation of 4-nitrophenyl-2-[18F]fluoropropionate.20 This tracer has been developed by an optimization strategy in which sugar moieties are introduced to improve the pharmacokinetics of the peptide. In murine tumor models as well as in patients, this radiopeptide showed receptor-specific tumor uptake and good clearance kinetics, resulting in high contrast images that noninvasively demonstrate αvβ3 expression; quantification with 18F-labeled RGD peptides is feasible.20,205–209
However, the multistep synthesis of 18F-labeled peptides using activated esters is complicated and time consuming. Chemoselective 18F-labeling strategies based on oxime formation using 4-[18F]fluorobenzaldehyde210,211 or with [18F]fluorosilyl benzaldehyde212 have been introduced. The synthon 4-[18F]fluorobenzaldehyde has also been applied to the labeling of HYNIC-modified RGD precursor resulting in an hydrazone-linked RGD peptide.213 In a similar approach, an aminooxy-functionalized double-bridged RGD peptide has been conjugated to a series of 18F-labeled aldehydes, leading to [18F]-AH111585, which has already entered clinical trials.214 Other chemoselective strategies use thiol-reactive groups, including 18F-labeled glycosyl thiosulfonate (Ac3-[18F]FGlc-PTS) as a thiol-reactive glycosyl donor for glycosylation of peptides.215 This approach allows introduction of both the radiolabel and a pharmacokinetic modifier in one synthesis step. Another example for a thiol-reactive synthon is the maleimide 18F-FBEM, that has been applied to the labeling of monomeric and dimeric thiolated RGD peptides.216 The advantages of the so-called “click” chemistry have been applied to radiopharmaceutical chemistry, to take advantage of its selectivity, reliability, and speed under aqueous mild Cu(I)-promoted reaction conditions. Following this approach, a comparison of different strategies of chemoselective labeling of alkyne-functionalized RGD peptides confirmed that “click labeling” of peptides could represent an attractive strategy.217 Furthermore, applying the click-chemistry–based glycosylation method for 18F-labeling, it was shown that the product [18F]FGlc-RGD could be synthesized in a significantly improved radiochemical yield in comparison with [18F]Galacto-RGD, when successful imaging of αvβ3-integrin expression in vivo suggested high potential of [18F]FGlc-RGD for future application in PET imaging studies.69 In addition, [18F]RGD-K5, which is another RGD-based PET tracer synthesized by Cu-catalyzed azide–alkyne coupling, has already entered initial clinical trials.218
Positron-emitting radiometals such as 64Cu and 68Ga have become of increasing interest for the labeling of peptides, because these radionuclides do not require an on-site cyclotron. Many studies have focused on the development of metal-labeled chelator-bearing RGD peptides. The peptide DOTA-RGDyK has been labeled with 64Cu and shows a high tumor/blood and tumor/muscle ratios in vivo of approximately—seven to eight using small-animal PET imaging. High uptake was observed also in liver, intestine, and bladder requiring further structural optimization of the tracer.219 Other RGD peptides reveal a tendency to bind to blood proteins resulting in higher blood pool radioactivity in vivo.220
Jeong et al.221 used isothiocyanatobenzyl-1,4,7-triayzacyclononane-1,4,7-tiracetic acid (SCN-Bz-NOTA) and conjugated it with c(RGDyK) for small-animal PET imaging of mice-bearing SNU-4C xenografts which showed adequate tumor visualization. In initial patient studies, the NODAGA chelating system was conjugated to c(RGDfK). The resulting [68Ga]NODAGA-RGD clearly demonstrated decreased protein-binding properties in vitro and improved blood clearance in vivo compared to [68Ga]DOTA-RGD.24 The improved imaging properties and the straightforward radiosynthesis make [68Ga]NODAGA-RGD an interesting potential alternative to 18F-labeled RGD peptides to image αvβ3 expression.
MULTIMERIC TRACERS: RADIOCHEMICAL STRATEGIES
The so-called multimerization approach means that more than one binding epitope is included in the targeting molecule resulting in increased ligand concentration.
A glutamic acid linker has been used for the synthesis of a dimeric RGD peptide when coupling two cyclo(-RGDfK) and DOTA or HYNIC for radiolabeling.222,223 The dimer 99mTc-HYNIC-E-[c(RGDfK)]2 revealed a 10-fold higher affinity for αvβ3 and an improved tumor retention but also a higher uptake in kidneys compared with the monomeric 99mTc-HYNIC-c(RGDfK). When a series of monomeric, dimeric, tetrameric, and octameric RGD peptides linked via PEG moieties was studied,210,211,224 increasing binding affinities with larger peptide mass has been found in vitro and confirmed in vivo by PET imaging.
18F-labeled glutamic acid–bridged dimeric RGD peptide ([18F]FB-E[c(RGDyK)]2) improved tumor uptake and retention compared to the monomer. Interestingly, the dimer had predominant renal excretion, whereas the monomer was excreted via the biliary route.225,226 Very similar effects have been reported on the multimeric 64Cu-labeled analogs,227 when the tetrameric [64Cu]DOTA-E[E-c(RGDyK)2]2228 showed superior integrin-binding affinity with rapid and improved tumor uptake in vivo in comparison with the monomeric and dimeric RGD analogs. The octameric RGD peptide further confirmed this finding229; however, the tracer uptake in the kidneys and muscle was increased, suggesting that a balance between binding epitope density and tracer size is important for the design of the optimal tracer. Recently, studies on regio-selective addressable functionalized templates (RAFTs)230 or dendrimers231 as a scaffold for the synthesis of multimeric RGD peptides have been reported. In the case of the [99mTc]RAFT-RGD, four cyclic RGD sequences are tethered on a cyclodecapeptide platform. Very recently, Wängler et al. reported on the synthesis of the first cyclic RGD hexadecimer using polyamidoamine (PAMAM) dendrimers for radiolabeling with 68Ga, when the obtained multimeric systems were conjugated to a new DOTA-based chelator developed for the derivatization of sterically demanding structures. As expected, the multimers showed very high avidities in vitro—increasing with the number of RGD moieties of up to 130-fold compared to the monomer.
The series of multimeric RGD systems are promising tracers to study their in vivo pharmacodynamic properties with small-animal PET and to develop optimized tracers with improved in vivo pharmacokinetics and high retention in the tumor.
RADIOLABELED RGD PEPTIDES: CLINICAL USE
The emerging field of molecular imaging of angiogenesis by PET is of utmost clinical importance and could significantly improve the assessment of the response to antiangiogenic treatment or combined cytotoxic/antiangiogenic therapy. Until now, data on the clinical value of PET imaging of angiogenesis has been scarce.
A variety of preclinical studies clearly indicate the potential value of PET imaging of αvβ3 expression for response assessment. Besides tumor-associated angiogenesis, the nontumor–associated pathologic states reflected by angiogenesis, such as physiologic changes in the bone metastatic microenvironment in arthritis research232 or PET imaging of microvessel density during low-dose paclitaxel therapy with [18F]AH111585 ([18F]fluciclatide) have been studied.21 Furthermore, the response of human glioblastoma xenografts to antiangiogenic sunitinib therapy has been monitored by [18F]fluciclatide-PET. A reduction of tumor microvessel density was identified earlier than any significant volumetric changes.233 Similarly, the antiangiogenic agents ZD4190 or Sunitinib significantly reduced the uptake of [18F]fluciclatide compared to untreated U87MG xenograft tumors from day 2 onward post therapy initiation.234,235
To date, the initial clinical trials with [18F]fluciclatide have demonstrated that this radiopeptide can successfully image metastatic breast cancer lesions in vivo. Kenny et al. studied seven patients with a total of 18 tumors detectable by computed tomography with [18F]fluciclatide PET. All tumors were detected by the PET tracer.236 Increased uptake compared with background was demonstrated in metastases in lung, pleura, bone, lymph node, and primary tumor. The authors concluded that [18F]AH111585 is safe, metabolically stable, and able to detect breast cancer lesions by PET at most anatomic sites. Further clinical trials with [18F]fluciclatide are in progress.237,238
Most clinical data involving angiogenesis imaging involves [18F]Galacto-RGD. [18F]Galacto-RGD can successfully image αvβ3 with good tumor/background ratios.206 The biodistribution of the tracer in patients is characterized by rapid, predominantly renal tracer elimination, resulting in low background radioactivity in most regions of the body. A high inter- and intraindividual variance in tracer accumulation in tumor lesions was noted, suggesting substantial heterogeneity of αvβ3 expression. The effective imaging dose in patients was found to be similar to an [18F]FDG scan.208 The distribution volume values (Dv), which reflect the receptor concentration in tissue, were four times higher for tumor tissue than for muscle tissue, suggesting specific tracer binding. In a PET imaging study including 19 patients with solid tumors, [18F]Galacto-RGD uptake values were significantly correlated with the microvessel density and αvβ3 expression that have been determined by immunohistochemistry.207
Looking closely at various tumor entities, it was found that in squamous cell carcinoma of the head and neck (SCCHN) good tumor/background ratios were found, in accordance with immunohistochemical analysis that found predominantly vascular αvβ3 expression, suggesting that [18F]Galacto-RGD PET might be used as a surrogate parameter of angiogenesis in SCCHN.209 In patients with glioblastoma, a maximum tracer uptake was found in the highly proliferating and infiltrating areas of tumors. Immunohistochemical staining was prominent in tumor microvessels as well as glial tumor cells. In areas of highly proliferating glial tumor cells, tracer uptake in the PET images correlated with immunohistochemical αvβ3 expression of corresponding tumor samples.239
In an initial clinical study with [18F]RGD-K5 the tracer was administered to 12 patients with breast cancer, who also underwent [18F]FDG PET imaging.22 157 lesions were identified by [18F]FDG PET, of which 122 showed elevated [18F]RGD-K5 uptake. In a few cases (4%), [18F]RGD-K5 uptake was higher than [18F]FDG. Comparable to [18F]Galacto-RGD, [18F]RGD-K5 uptake in lesions did not correlate with [18F]FDG uptake. Biodistribution and radiation dosimetry was performed in four patients,240 with an estimated effective dose of 0.015 mSv/MBq when using a 1-hour bladder voiding interval, comparable to other integrin-targeting PET tracers. Recently, a nonrandomized, uncontrolled pilot phase II clinical study was initiated to assess the usefulness of [18F]RGD-K5 PET imaging to predict efficacy and early response monitoring of combination therapy with the anti-VEGF antibody, bevacizumab, plus chemotherapy.241
Clinical studies that compare [68Ga]NOTA-RGD and [18F]FDG for imaging of hepatic metastases of colorectal cancer before a combination therapy of FOLFOX and bevacizumab have been reported.242 Static PET images with 68Ga-NOTA-RGD were acquired 30 minutes after injection. Interestingly, the patients with 68Ga-NOTA-RGD uptake in the hepatic metastases showed partial response to the antiangiogenic combination therapy, whereas the other patients did not benefit from treatment.
Recently, the safety, biodistribution, and dosimetric properties of a dimeric RGD peptide (18F-FPPRGD2) have been studied by Mittra et al. in five healthy volunteers.23 This tracer has desirable pharmacokinetic and biodistribution properties with an acceptable effective radiation dose of 15 mSv for one PET scan. This dimeric RGD peptide is a promising candidate for PET evaluation of oncologic patients especially those with brain, breast, or lung cancer.
In summary, these studies report encouraging findings and suggest that PET imaging with radiolabeled RGD peptides, alone or in combination with additional functional imaging modalities, might help in patient selection for antiangiogenic therapies.
TRACERS FOR THE VASCULAR ENDOTHELIAL GROWTH FACTOR AND ITS RECEPTORS
VEGF and its two receptor subtypes, Flt and KDR, are the most important regulators of angiogenesis and are key molecular targets for imaging or anticancer treatment.243,244 VEGF is a dimeric, disulfide-bound glycoprotein, with at least seven homodimeric isoforms that differ in their biologic properties such as the ability to bind heparin245: VEGF165 (with a heparin-binding domain), freely soluble VEGF121 (without heparin-binding domain) or VEGF189 and VEGF206 that remain localized to the ECM.246 The increased expression of VEGF by tumor cells and VEGFR-2 (KDR) and VEGFR-1 (Flt) by the tumor endothelia correlate with proliferation, microvessel density, tumor metastatic potential, and poorer patient prognosis in a variety of malignancies.247 More recently, the approval of the humanized anti-VEGF monoclonal antibody bevacizumab (Avastin) for first-line treatment illustrates the important role of the VEGF pathway for tumor therapy.245 Bevacizumab inhibits VEGF-induced endothelial cell proliferation, permeability, and survival.248,249 The development of specific and selective molecular imaging probes for a noninvasive imaging modality would significantly facilitate the assessment of several parameters, such as the occupancy of VEGFRs in vivo, for a sophisticated approach toward individual therapy.250
Antibodies Against VEGF
One category of tracers to track VEGF expression are antibodies against VEGF itself. VEGF is highly expressed in many human tumor types.26 89Zr-labeled bevacizumab has been used in small-animal PET imaging studies, showing significantly higher tumor uptake compared to human 89Zr-IgG in an ovarian tumor xenograft model.26 Comparable results were reported for an yttrium-labeled DTPA conjugate of bevacizumab251 and the 124I-labeled monoclonal antibody VG67e.252 The humanized form of the mouse monoclonal anti-VEGF antibody MV833 was labeled with 124I-iodine and the biodistribution of HuMV833 in patients in a phase I clinical trial demonstrated that this radiolabeled antibody is a promising new tracer for in vivo imaging of VEGF in the tumor microenvironment. 27 However, the antibody distribution and clearance properties were quite heterogeneous not only between and within patient groups but also between and within individual tumors.27 Furthermore, correlation of tracer uptake to the level of VEGF expression in the tumor tissue was not always confirmed.253 Penetration of the radiolabeled antibody into the tumor is hampered by the large size of the antibody resulting in days before a specific imaging signal could be obtained. Efforts have been reported that combine the use of smaller antibody fragments with easy-to-label NOTA conjugates to overcome this problem.254
More recently, 89Zr-bevacizumab PET imaging of early antiangiogenic tumor response to treatment with an HSP90 inhibitor has been successfully performed.255 Moreover, 89Zr-labeled ranibizumab has been used to monitor changes in the tumor during Sunitinib treatment. PET imaging of this tracer was compared to [18F]FDG and 15O-water PET.256 89Zr-labeled ranibizumab successfully demonstrated dynamic changes during sunitinib treatment within the tumor. The PET results corresponded with tumor growth and immunohistochemical vascular and tumor markers demonstrating that PET imaging allows serial analysis during treatment studies.256
Radiolabeled VEGF Derivatives
A second category of tracers includes radiolabeled VEGF and VEGF derivatives to image VEGF receptor expression in vivo.257 Both VEGF165 (with a heparin-binding domain) and VEGF121 (without heparin-binding domain) have been used for VEGFR imaging.29,258 There is high retention in the kidney, which is the dose-limiting organ that expresses high levels of VEGFR-1.259 Compared to VEGF121, VEGF165 is less soluble because of the presence of its extra domain for heparin binding. This results in increased nonspecific binding and a low tumor-to-background ratio. Therefore, VEGF121 is in principle the more suitable lead structure for the development of radiolabeled VEGF analog PET tracers compared to VEGF165.
Using highly vascularized tumor xenografts that overexpress VEGFR-2, 64Cu-labeled VEGF121 revealed rapid, specific, and prominent uptake in the tumor region on small-animal PET imaging.29,260 However, the problem of VEGFR-1–mediated high uptake in the kidney was manifested and addressed.29,258,261–263 The distinct domains of VEGF that interact with VEGFR-2 and VEGFR-1 have been identified by alanine-scanning mutagenesis, revealing that Arg82, Lys84, and His86 are critical for the affinity to VEGFR-2, whereas Asp63, Glu64, and Glu67 are required for the binding of VEGF to VEGFR-1.264 Based on this observation, the D63AE64AE67A mutant of VEGF121(VEGFDEE) was conjugated with DOTA and labeled with 64Cu (64Cu-DOTA-VEGFDEE) to aim at selective PET imaging of VEGFR-2 expression.265 Compared to 64Cu-DOTA-VEGF121, 64Cu-DOTA-VEGFDEE had comparable tumor-targeting efficacy but reduced renal accumulation. Further improvement in VEGFR-2–binding affinity, specificity, pharmacokinetics, and tumor-targeting efficacy by designing alternative VEGF121 mutants with explicit receptor subtype selectivity was considered advantageous for clinical translation of suitable VEGF-based imaging probes for PET. In another study, 64Cu-DOTA-VEGF121 has also been used to image experimental myocardial infarction, demonstrating the feasibility of imaging increased VEGF receptor expression in vivo in preclinical models.266 However, the VEGF-based imaging agents in general suffer from awkward biodistribution, rendering these tracers unsuitable for human applications because of the unacceptably high tracer uptake in liver and kidney.258,263,267–269 Significant progress has been achieved by the development of a single-chain Cyc-tagged VEGF (scVEGF), that has been labeled with 99mTc directly.28 It was also used for labeling with 64Cu when a PEG linker and a DOTA chelator were conjugated.262 This tracer showed considerably decreased kidney uptake and higher tumor uptake when compared to the analogous 99mTc-labeled HYNIC-conjugate of scVEGF.
PET imaging of VEGF receptor expression in vivo could evolve to be a valuable tool for treatment planning, and monitoring of therapy response,265,266,270 particularly in antiangiogenic therapies that block VEGF/VEGFR-2 function.271
TRACERS FOR MATRIX METALLOPROTEINASES
The MMPs are secreted by and expressed on the cell surface of various cell types and they are capable of degrading proteins of the ECM.272 The MMP family consists of more than 25 proteases that are divided into five subgroups: Collagenases (MMP-1, -8, -13), gelatinases (MMP-2, -9), stromolysins or matrilysins (MMP-3, -7), membrane-type MMPs (MMP-14 to -17), and other nonclassified MMPs.273 The MMPs play an important role in various physiologic and pathologic processes such as angiogenesis, tissue repair, inflammation, including the assessment of atherosclerotic plaque vulnerable to rupture, and tumor development.274,275 Their effects and enzyme activity are controlled by a balance between the expression of endogenous MMP inhibitors and proenzyme synthesis,276 when the increased proenzyme production results in the degradation of the basement membrane and the extracelluar matrix.277 The MMP-2 and -9 are most commonly expressed in malignant tissue, emphasizing their important role in tumor-induced angiogenesis and metastasis,187,278–280 where their occurrence is correlated with the aggressiveness and metastatic potential of tumors. Therefore, MMPs are potential molecular targets for radionuclide imaging studies and therapeutic interventions.25,281,282 Numerous efforts have been made to develop radiolabeled MMP inhibitors that are suitable for these purposes. Many of them are in preclinical or clinical studies.
Peptide-Based Tracers for Matrix Metalloproteinases
Starting from a phage display library approach, the decapeptide CTTHWGFTLC (CTT peptide) was identified to selectively inhibit MMP-2 and -9.283 This peptide suppressed the migration of endothelial and tumor cells in vitro. Moreover, it was shown that the peptide mediated the homing of phages in the tumor vasculature in vivo. The derivatization of this peptide at the N-terminal end by tyrosine did not affect the inhibitory capacities for MMP-2 binding significantly, and, most importantly, the radioiodinated peptide was stable toward degradation by activated MMP-2 and -9.284 However, the evaluation of this tracer revealed low metabolic stability in vivo and high lipophilicity, rendering this tracer unsuitable for in vivo imaging studies. The CTT peptide was also conjugated with DOTA and labeled with Cu-64.285 In this case, there has been some evidence for selective uptake of [64Cu]DOTA-CTT by gelatinase-expressing tumors. However, the low affinity for MMP-2 and -9 and its marginal stability in vivo make this bioconjugate an inadequate radioligand for PET imaging studies of MMP expression. More recently, peptide-based imaging probes have been designed.286–288 Ujula et al.286 compared three structurally diverse [68Ga]DOTA-conjugated peptides derived from TCTP-1 to target MMP-9, including two cyclic peptides bearing a cystine bridge and a lactam bridge, respectively, and a linear peptide. The results did not provide a significant progress, because the authors conclude that further modifications of the tracer are needed to improve the stability and affinity of the peptides. In another study, a dual-labeled peptide imaging agent was utilized that targets MMP-2/9 and is labeled with an infrared dye and 68Ga.287 However, the 68Ga complexation greatly reduced binding affinity of the peptide which caused the loss of the PET signal, whereas the near-infrared imaging showed a specific fluorescent signal that clearly correlated to MMP-9 expression.287
Very recently, Chuang et al. used the PEGylation of the peptide GPLGVR with subsequent linkage to a tetramethylrhodamine (TMR) domain and 18F-labeling by N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB). Using small-animal PET, these authors successfully demonstrated that this 18F-labeled peptide showed high selectivity to image MMP expression in vivo. It was concluded that this strategy could be extended to other proteases and could be applied to the prediction of patient prognosis in future studies.288
Small Molecular Mass Tracers for Matrix Metalloproteinases
A large series of numerous nonpeptide inhibitors are currently being investigated as tracers for MMP. Detailed structure activity investigations revealed the most important structural parameters that determine high affinity binding289–292: (a) The coordination site binding to the catalytic zinc ion, (b) a lipophilic site interacting with a hydrophobic cleft, and (c) hydrophobic interactions between the sulfonylamide substituent of the ligand and the binding pocket.
Early in these approaches, 18F-labeled MMP-2 inhibitors have been derived from D-amino acid scaffolds,293 when in vitro studies demonstrated micromolar inhibitory activities. A broad range of MMP inhibitors belonging to the N-sulfonylamino acid family have been labeled with 11C and 18F, mostly at the phenyl group or at the hydroxamic acid moiety, and evaluated by their inhibitory effectiveness for the gelatinases MMP-2 and -9.294–298 In addition, radiolabeled biphenylsulfonamides have been developed, which showed high inhibitory effectiveness on MMP-13.299 Oltenfreiter et al.300,301 studied hydroxamic and carboxylic acid containing MMP inhibitors that were derived from amino acid scaffolds and labeled by 123I-iodine. Consistently, these candidates also showed high inhibitory capacities on gelatinases in vitro. Moreover, a series of fluorinated derivatives from the sulfonylamide type has been systematically studied, when a promising hydroxamic acid derivative was identified and labeled with 18F.302 This approach was further extended by the introduction of 18F-radiolabeled nonhydroxamate MMP inhibitors, where the effect of PEGylation on MMP subtype selectivity and affinity was studied in detail.303
Contrary to the promising in vitro data, the in vivo imaging studies with these radiotracers have not yet been fully elucidated or were disappointing. Although the biodistribution data of some tracers indicated favorable pharmacokinetics in normal mice,298,300,301,304 further micro-PET imaging studies using murine tumor models demonstrated negligible tracer accumulation in the tumors.296,305,306 More recently, a shelf-stable arylboronic ester of Marimastat, a nonselective MMP inhibitor, was successfully used for 18F-labeling to give [18F]marimastat-aryltrifluoroborate and subjected to in vivo experiments using 67NR mammary carcinoma-bearing mice, when localization of the tumor by PET imaging was shown.307
For SPECT imaging, RP782, a macrocyclic hydroxamic acid originally developed by Bristol-Myers Squibb Medical Imaging on the basis of the previously reported structures of MMP inhibitors, has been labeled with 111In and successfully applied to image vascular remodeling and aortic macrophage content.308–310 Molecular imaging of MMP activation remains a useful and promising tool, especially for tracking plaque biology and response to therapy in the field of atherosclerosis research.
ED-B DOMAIN
Biologic Background
Fibronectin is an ECM protein that exists in several isoforms (e.g., III CS, ED-A, ED-B) and is involved in a variety of processes including wound healing, cell migration, and oncogenic transformation.311 It has been found that the ED-B domain containing fibronectin isoform plays an important role in vascular proliferation and is widely expressed in fetal and neoplastic tissues, whereas its distribution is highly restricted in normal adult tissue.312 In a murine tumor model, a fluorescent-labeled anti–ED-B single-chain Fv antibody fragment selectively accumulates around blood vessels of the tumor vasculature.313
Development of Tracer Targeting the ED-B Domain
Tracer development started with the introduction of a radioiodinated anti–ED-B antibody fragment with picomolar binding affinity. This single-chain fragment (scFv(L19)) accumulated in several murine tumor models.30 In addition, microautoradiography and immunohistochemical staining showed selective accumulation in tumor vasculature whereas no activity accumulated in organ vessels. An initial clinical study using the 123I-labeled dimeric single-chain fragment L19(scFv)2 and SPECT that included patients with brain, lung, or colorectal cancer demonstrated, for 16 out of 20 patients, different levels of tracer accumulation either in the primary tumor or metastases 6 hours p.i.314Besides the 123I-labeled derivative, 99mTc-labeled compounds have been developed for SPECT imaging.315 L19 was either genetically modified expressing a (Gly)3-Cys-Ala (AP39) or a (His)6 tag (L19-His) sequence at the C-terminal end or chemically modified by introducing a MAG2-type chelator at amino functions of lysine side chains (L19-Hi20). Labeling was carried out either via 99mTc-pertechnetate (AP39, L19-Hi20) or the 99mTc-tricarbonyl method (L19-His). 99mTc-AP39 had the best imaging properties including highest tumor uptake and predominantly most rapid renal excretion.
A variety of different antibody formats have been evaluated to identify a radioimmunoconjugate best suited for imaging.316,317 The complete human IgG1 L19-IgG1 (∼150 kDa), the “small immunoprotein”L19-SIP (∼80 kDa), and the single-chain fragment scFv(L19) (∼50 kDa) were included. The single-chain fragment had the most rapid clearance but also the lowest metabolic stability. The elimination kinetics for the single-chain fragment scFv(L19) was not compatible with the short half-life of the PET radionuclides usually used for labeling peptides such as 18F and 68Ga. Therefore, PET radionuclides with longer half-lives like 76Br (half-life 16.2 hours) and 124I (half-life 4.18 days) have been used to develop radiolabeled antibody fragments which allow PET imaging of the ED-B domain expression using PET. In one approach the 80-kDa L-19-SIP was labeled with 76Br via enzymatic radiobromination. The serum stability of 76Br-L-19-SIP was comparable to the 125I-labeled analog but tumor/blood ratio was not higher than 1.2 even after 24 hours p.i. resulting in high background activity in a murine tumor model.31 Subsequently, 124I-L19-SIP was developed for immuno-PET imaging and guidance of 131I-L19-SIP radioimmunotherapy.318 Labeling was carried out with a GMP-compliant production of 124I-iodine and using the IODOGEN method. The 124I-labeled compound demonstrated comparable tumor/background ratios as found for 131I-L19-SIP and better ratios compared to the 76Br-labeled analog. At 72 hours after tracer injection, good tumor/background ratios were found for most organs in a murine tumor model. Lowest ratios were found for colon (5.3), ileum (5.5), and bladder (8.6).
Despite these promising results further studies are necessary to determine if these tracers are suitable for imaging the ED-B domain.
RECEPTOR IMAGING AND RECEPTOR-MEDIATED THERAPY
Based on the successful introduction of radiolabeled somatostatin derivatives in receptor scintigraphy and PRRT, other receptors have been studied as targets. This include the estrogen receptor, the chemokine receptor CXCR4, the melanocortin-1 receptor, the neurokinin type 1 receptor, the CCK2/gastrin receptor, the neurotensin receptors, the bombesin receptors, and receptors for the vasoactive intestinal peptide (VPAC1 and VPAC2).
Somatostatin Receptor
A variety of malignant tumors, including NETs, small-cell lung cancer, breast tumor, and malignant lymphoma overexpress SSTRs. This receptor class is divided into five subtypes (SSTR1–SSTR5) of which subtype 2 is predominantly expressed on tumors. SSTRs regulate a great variety of diverse physiologic processes including hormone secretion, neuromodulation, smooth-muscle contractility, nutrient absorption, apoptosis, and cell growth.319,320Radiolabeled somatostatin–derived analogs are currently used for diagnoses and treatment of SSTR-positive tumors in nuclear medicine.321
Naturally occurring somatostatin exists in two isoforms (14 amino acids and 28 amino acids); both have short plasma half-lives (∼2 minutes), which makes them unsuitable as imaging probes. Thus, the development of imaging tracers is focused on derivatives of octreotide, a disulfide-bridged eight amino acid analog of somatostatin.
The first routinely used radiotracer for SSTR planar and SPECT imaging was 111In-pentetreotide, an 111In-labeled DTPA-conjugated octreotide derivative.32 This radiopharmaceutical, however, has several drawbacks, mainly related to the use of 111In including limited availability, high isotope costs, and a medium γ-energy leading to suboptimal image resolution and relatively high patient radiation burden. Nevertheless, because of the commercial availability, without the need for in-house radiopharmacy, this radiopharmaceutical is still widely used to image SSTR expression. An alternative planar and SPECT tracer is 99mTc-EDDA/HYNIC-TOC.33 This compound can be prepared using a kit procedure in high radiochemical purity and yield. A comparison of both tracers in a clinical study demonstrated better tumor-to-background ratios and higher sensitivity for 99mTc-EDDA/HYNIC-TOC compared to 111In-pentetreotide.322
With the increasing availability of 68Ga-generators323 and the superior imaging quality of PET, 68Ga-labeled octreotide derivatives have become more and more routine for imaging. The most commonly used PET tracer for the diagnosis of SSTR-expressing tumors is [68Ga]DOTA-TOC.324 The synthesis is straightforward and includes elution of the generator either via the fractionated elution approach34 or the preconcentration technique,325 heating of the 68Ga-chloride/peptide/buffer mixture, and subsequent purification using a SEPPAK cartridge. A variety of remotely controlled systems are available to carry out this labeling process.323,326 Alternative tracers for PET imaging of SSTR-positive tumors include [68Ga]Lanreotide,327 [68Ga]DOTA-TATE,35 and [68Ga]DOTA-NOC.36 Labeling of the different compounds is comparable with small differences in radiolabeling yield. Some differences are found in the tracer affinity for the different SSTR subtypes. Whereas DOTATOC and DOTATATE predominantly bind to subtype 2, a broader binding profile is found for the other octreotide derivatives.38
Some studies have been carried out to develop 18F- and 64Cu-labeled derivatives for PET imaging of SSTR expression. Although 64Cu is an attractive PET radionuclide, few studies have been performed. 64Cu-DOTA-TOC328 and 64Cu-TATE-OC329 have been described with promising imaging properties in preclinical studies. Labeling is straightforward: The cyclotron produced 64Cu is incubated with the corresponding precursor in buffer solution and subsequently purified using a SEPPAK cartridge. Limitations include the restricted availability of 64Cu and the lack of clinical data confirming the utility of these compounds in clinical settings. Besides 64Cu, 18F-labeled octreotide derivatives have been developed.330 In general, 18F-labeling of peptides has to be carried out using prosthetic group labeling approaches, which are inferior for labeling of peptides.
Most of the development of octreotide derivative radiotracers employ SSTR agonists. The uptake mechanism is characterized by specific binding to the corresponding SSTR subtype, internalization via endocytosis, release of the radiotracer in the cell, and finally, metabolism resulting in radiolabeled fragments, which cannot penetrate the cell membrane and thus accumulate.331 The major paradigm was that internalization is essential for high tracer accumulation. However, in the last few years, antagonists have been developed. Despite the “internalization paradigm,” 111In-DOTA-SST3-ANT and 111In-DOTA-SST2-ANT demonstrated higher receptor targeting in a nude mouse model than the agonist 111In-DTPA-TATE.332 Most recently, in an initial clinical study, using 111In-DOTA-SST2-ANT (111In-DOTA-BASS), imaging of NETs with SSTR antagonists resulted in even higher tumor uptake and better visualization of metastatic NETs than the reference agonist 111In-DTPA-octreotide.37 However, further studies are necessary to determine if these antagonistic radiopharmaceuticals will become attractive alternatives to the currently established agonists.
The great benefit of this SSTR-targeting radiopharmaceuticals is that they can be used for peptide receptor–mediated radionuclide therapy. The major preparations are [90Y]DOTA-TOC and [177Lu]DOTA-TATE.38 Tracer selection is mainly based on tumor burden/size. Patients with higher tumor burden were initially treated with [90Y]DOTA-TOC (β−-energy of 90Y: 2.3 MeV) and patients with smaller tumors with [177Lu]DOTA-TATE (β−-energy of 177Lu: 0.5 MeV). The higher nephrotoxicity of [90Y]DOTA-TOC can also have an influence on treatment planning. Recently, it was found that combined treatment with both isotopes has a favorable treatment outcome.333
Radiolabeling of both classes of compounds is straightforward. The peptide precursor is incubated for approximately 30 minutes with the corresponding radionuclides (177LuCl3 or 90YCl3) in the presence of an ascorbic acid buffer at either 80° or 95°C (temperature differs depending on the radionuclide used). A variety of remote-controlled systems have been evaluated for the production of this radiopharmaceuticals allowing automated synthesis and reducing personnel radiation exposure.334 Additional radiopharmaceuticals used in SSTR-mediated radionuclide therapy are [90Y]DOTA-TATE, [90Y/177Lu]DOTA-NOC, and [90Y]DOTA-LAN.38 Radiolabeling is, in general, similar to [90Y]DOTA-TOC and [177Lu]DOTA-TATE.
OTHER TARGETS
Estrogen Receptor Imaging
Estrogen receptor expression is an important indicator of breast cancer behavior and is a critical determinant of the response of endocrine therapies. Two different forms of the receptor are found: Estrogen receptor, α and β. Because of hormone activation, estrogen receptors form homo- and heterodimers.335 Estrogen receptors are overexpressed in several tumors including ovarian, prostate, and especially breast carcinoma.336 There are two hypotheses to explain the role of these receptors in the development of breast cancer336: (a) Binding of estrogens to the estrogen receptor stimulates proliferation of mammary cells, increasing the target cell number within the tissue. The resulting increase in cell division and DNA synthesis elevates the risk for replication errors, which may result in mutations that disrupt normal cellular processes (e.g., apoptosis, cellular proliferation, or DNA repair); (b) estrogen metabolism leads to the production of genotoxic by-products that could directly damage DNA. One ligand binding to both estrogen receptors (ERα and ERβ) with equivalent affinity is 17β-estradiol. This is the lead compound for the development of a radiotracer to image estrogen receptor expression with PET.337
Almost 30 years ago, studies were carried out to compare 17β-estradiol derivatives that were 18F-labeled at different positions.39 18F-16α-17β-flouroestradiol ([18F]FES) was the most promising tracer with a 6.3-fold preference for ERα.338 Although a variety of other radiotracers have been developed and tested.337 [18F]FES remained the most successful and the only one in routine use to image estrogen receptor expression. Although some compounds, such as 18F-labeled moxesterol,339 showed superior properties compared to [18F]FES in preclinical studies, they have not been successful in human studies. The original synthesis described by Kiesewetter et al.,39 which is based on the nucleophilic fluorination of the corresponding triflate, was optimized by different groups resulting in a synthesis procedure that allowed automated synthesis of the radiopharmaceutical with a decay-corrected yield of approximately 30% after 60-minute EOB.337
Chemokine Receptor CXCR4
The chemokine receptor CXCR4 is a key player in the process of cancer metastasis formation.340 Interaction of CXCR4 with its ligand CXCL12 leads to upregulation of urokinase-type plasminogen activator receptor (uPAR) and metalloproteinases (especially MMP-2 and -9 which increase tumor invasiveness and tumor-induced angiogenesis.341 The expression of chemokine receptors on tumor cells is not random. CXCR4 is the most commonly expressed chemokine receptor on tumor cells. CCR7, another commonly expressed chemokine receptor on tumors, is mainly responsible for metastasis to the lymph nodes. The significance of binding of the ligand CXCL12 to CXCR4-expressing tumors is unclear.
Because of the central role in metastasis formation, CXCR4 is an interesting target for radiotracer development. The target structures include small molecules, peptides, and antibodies.342 One nonpeptide CXCR4 inhibitor already in clinical trials is the bicyclam AMD3100. Because of its cyclam rings, it was labeled with 64Cu.40 This tracer can be used to determine increased CXCR4 expression in metastasis in murine tumor models. However, further clinical studies are necessary to determine the full potential of this tracer as well as on a monocyclam derivative radiolabeled with 18F-fluorine.343
In 1998, T140, a disulfide-bridged 14-residue peptide was identified to be a potent CXCR4 antagonist.344 Based on this structure, a variety of modifications mainly to improve serum stability and to allow radiolabeling have been made resulting in 111In-, 18F-, and 64Cu-labeled derivatives.342 To date, the data yet seems not to be very promising. Further downsizing of the molecule leads to a cyclic pentapeptide FC131,345 which is the basis of the development of 68Ga-labeled peptides to target CXCR4.41,42 Initial data suggest high whole-body contrast with fast renal clearance and low liver uptake. None of these tracers are in clinical trials.
Radiolabeled α-MSH Derivatives for Melanoma Imaging
The melanocortin-1 (MC1) receptor is one member of five melanocortin receptors (MC1–MC5) belonging to the superfamiliy of G-protein–coupled receptors (GPCRs). MC1 is primarily involved in regulation of skin pigmentation. Its interaction with α-melanocyte–stimulating hormone (α-MSH) peptide initiates a complex signaling cascade leading to the production of the pigment eumelanin.346 MC1 receptors are highly overexpressed on melanoma cells and α-MSH peptide analogs bind with nanomolar affinities to the MC1 receptor making radiolabeled α-MSH derivatives promising candidates as melanoma-specific imaging probes.43
Initial studies used radioiodine-labeled α-MSH derivatives. These derivatives, however, were unstable (dehalogenation) leading to limited use even for in vitro studies. One of the most widely used α-MSH derivatives is Ac-[Nle4, D-Phe7]-α-MSH (NDP), which is referred to as the gold standard and used as a lead structure for tracer optimization because of the high receptor affinity and the metabolic stability.347 Introduction of prosthetic groups like N-succinimidyl-3-iodobenzoate (SIB) or N-succinimidyl-4-iodobenzoate (PIB) further improves stability. Additional optimization is based on conjugation of the linear α-MSH derivatives with different chelating systems like DTPA, DOTA, NxSy-systems, and pyrazolyl-diamine backbones to enable labeling with 111In and 99mTc (including the 99mTc(CO)3 system). Although stability could be increased, tumor uptake and tumor retention remained poor.44Introduction of structural constraints was a further approach toward improved α-MSH derivatives. Strategies include cyclization via metal coordination,348 disulfide,349 and lactam350 formation. For example, 111In-DOTA-Re(Arg11)CCMSH, where rhenium coordination induced the constraint structure, yielded good imaging properties in a melanoma mouse model.46
Most efforts in the development of PET tracer for melanocortin-1 receptor imaging were focused on 64Cu45,351 and 68Ga-labeled α-MSH352,353 derivatives. A truncated form of NDP (NAPamide) was conjugated with DOTA for labeling with both isotopes, which showed clear tumor accumulation in a murine melanoma model.354 Although DOTA is not an optimal chelating system for 64Cu.351 Another approach used the rhenium-constrained DOTA-Re(Arg11)CCMSH to label with 64Cu, 68Ga, and 86Y.44 An alternative copper chelating system (CBTE2A) was introduced. [64Cu]CBTE2A-Re(Arg11)CCMSH demonstrated comparable tumor uptake and reduced liver activity compared to [64Cu]DOTA-Re(Arg11)CCMSH.45
(Arg11)CCMSH was also used for therapy labeled with 188Re using the rhenium for the introduction of the constraint as well as the β-emitting radioactive source.355 Recently, DOTA-Re(Arg11)CCMSH was used to introduce 177Lu.356 In initial murine melanoma models, some therapeutic effects were demonstrated.
Neurokinin Type 1 Receptor Imaging and PRRT
Neurokinin type 1 (NK-1) receptor belongs to the family of GPCRs found in both the central and peripheral nervous systems. Binding of substance P to the NK-1 receptor is associated with the transmission of stress signals and pain, endothelium-dependent vasodilatation and inflammatory reactions.357 NK-1 receptor antagonists have been studied in migraine, emesis, and psychiatric disorders. Other diseases in which the NK-1 receptor system is involved include asthma, rheumatoid arthritis, and gastrointestinal disorders. Besides its role in the central and the peripheral nervous system, the NK-1 receptor is consistently overexpressed on glioma cells and tumor vessels.358 For example, autoradiography studies found overexpression of the NK-1 receptor in 55 of 58 gliomas of WHO grades 2 to 4. This overexpression is the basis for the development of radiolabeled substance P analogs.
One tracer already in initial clinical studies is substance P conjugated with 1-(1-carboxy-3-carboxypropyl)-1,4,7,10-tetraazacyclododecane-4,7,10-triacetic acid (DOTAGA).47 This DOTAGA-substance P was labeled with 111In for dosimetry calculations and 90Y for therapeutic approaches. For patient application the radiopharmaceutical was injected via a stereotactically implanted catheter system either directly into the tumor or the resection cavity. Initial data suggest some disease stabilization or improved neurologic status. However, further studies are needed to demonstrate the effectiveness of such approaches. In a recent study, a substance P derivative was labeled with the α-emitting 213Bi.48 213Bi-DOTA-[Thi8, Met(O2)11]-substance P was studied in five patients. Targeted local radiotherapy with this tracer may represent an innovative treatment strategy for critically located malignant glioma.
An alternative lead structure is the synthetic chlorotoxin TM-601, a 36 amino acid neurotoxin, which has been labeled with 131I-iodine.49 The radiopharmaceutical was found to bind to malignant melanoma with high affinity and showed high tumor retention. An initial clinical study showed that the radiopharmaceutical is well tolerated and may have antitumoral effect.
Imaging and Therapy of CCK2/Gastrin Receptor–Expressing Tumors
Cholecystokinin (CCK) is one of the most widespread neuropeptides in the CNS.359 It is initially characterized as a 33 amino acid containing peptide, but seems to be present in a variety of different biologically active forms including CCK39, CCK33, CCK8, and CCK4; all derived from the same precursor peptide. The most abundant in the brain is CCK8 with the amino acid sequence H-Asp-Tyr-Met-Gly-Trp-Met-Asp-Phe-NH2.
The CCK2 receptor, one of the three CCK-binding receptors identified so far, is located in brain, stomach, pancreas, and gall bladder.360 In the gastrointestinal tract, activation of the receptor by gastrin stimulates gastric acid secretion. CCK2 is also known as a gastrin receptor. CCK2 receptor belongs to the superfamily of GPCRs. CCK2/gastrin receptors have been indentified on various tumor tissues with a high incidence on medullary thyroid carcinoma (whereas differentiated thyroid cancers do not express CCK2 receptors).361 Other cancers with high CCK2 expression are astrocytomas, small-cell lung cancer, and ovarian cancer. Less frequently, it is found on meningiomas, breast carcinomas, and ovarian adenocarcinomas.360
Radiolabeled peptides for the noninvasive detection of CCK2 expression is based on the C-terminal CCK receptor–binding tetrapeptide H-Trp-Met-Asp-Phe-NH2.360 With the exception of methionine, which can be replaced by amino acids like leucine or norleucine,362 this minimal binding sequence is very sensitive to modifications. The developed peptides are either based on CCK8 or minigastrin360 and include a variety of modifications to improve tracer properties, in particular the metabolic stability (e.g., introduction of D- or unnatural amino acids and cyclization).
One of the first minigastrin derivatives with good tumor-targeting properties is 111In-DTPA-MG0 but this tracer has unfavorable high renal retention.50 Kidney uptake could be reduced by the N-terminally truncated derivative MG11 (removal of the five glutamic acids) but this results in lower metabolic stability.51 To improve stability, cyclic derivatives of MG11 have been designed, which do not result in the desired optimization of the metabolic stability.52 Recently, dimeric minigastrin derivatives with improved tumor–background ratios, low kidney uptake, and improved metabolic stability compared to the monomeric form have been described.363 Further studies are needed to demonstrate the potential of multimeric minigastrin derivatives for imaging CCK2 expression. Nock et al.53 conjugated a tetraamine to the N-terminal end of different minigastrin derivatives for labeling with 99mTc and found high uptake in the kidneys, which could be reduced by co-injection of poly-Glu–containing peptides. By contrast, minigastrin derivatives including His tags showed drastically reduced kidney uptake.54
Initial studies with radiolabeled CCK8 derivatives demonstrated that nonsulfated CCK analogs are highly promising radiopharmaceuticals for imaging CCK2 expression.55 In murine tumor models, 99mTc-labeled sulfated CCK8 showed even a higher tumor accumulation than the nonsulfated derivative.56 Radiolabeled CCK8 combines good tumor targeting with low renal retention making this class of tracer good candidates to target CCK2 receptor.364
Initial clinical studies have been carried out with both gastrin- and CCK-like peptide derivatives (for review, see Roosenburg et al.360). Because of the different study protocols used comparison of the different approaches is difficult. Nevertheless, it was found that (a) imaging of CCK2-positive tumor is possible, (b) metabolic stability of the tracer in vivo is low, (c) 99mTc-Demogastrin 2 seems to be more stable than 111In-DOTA-MG11, and (d) although 90Y-DTPA-MG0 therapeutic responses were encouraging, renal toxicity in a few patients was rather severe. Altogether, further studies are needed to determine the most appropriate CCK2-targeting peptide for imaging as well as PRRT.
Radiolabeled Vasoactive Intestinal Peptide
The vasoactive intestinal peptide (VIP) is a neuroendocrine hormone that mediates a wide range of physiologic functions, including vasodilatory responses, growth and proliferation of normal and malignant cells. There are two receptor subtypes (VPAC1, VPAC2) that are expressed on a variety of NETs, breast and other adenocarcinomas, and small-cell lung cancer. VIP receptors are also expressed in the lung, gastrointestinal tract, peripheral blood cells, and kidney, which, to some extent, limit the use of this molecular target for imaging purposes. It is unlikely that VIP receptor expression imaging can be used to detect lymph node and liver metastasis, because the tumor-to-background ratio is poor. Nevertheless, intensive research studies proceeded to develop tracers based on the VIP peptide. VIP consists of 28 amino acids with a specific sequence required for full biologic activity.365 The sequence contains two tyrosines that provide an opportunity for labeling with radioactive iodine (e.g., 125I and 123I). 123I-iodine–labeled VIP has been studied in clinical trials by Virgolini et al.,57,366 who reported that 123I-iodine–labeled VIP and [111In]DTPA-DPhe1-Octreotide have a high sensitivity to locate tumor sites in 130 patients. These promising results for colorectal and breast cancer imaging have encouraged other research groups to develop VIP analogs labeled with 99mTc-technetium, 64Cu-copper and 18F-fluorine.367–369
Biodistribution studies of a VIP analog [99mTc]TP3654 in nude mice-bearing LS174T colorectal tumors showed high tracer uptake and high retention of radioactivity in the tumor (∼0.23%ID/g) at 4 to 24 hours after tracer injection.368,370 Albeit low, the tumor uptake was twofold higher than for [111In]octreotide and approximately four times higher than for [125I]VIP. In an initial study including 11 tumor patients, mainly with breast cancer, all known tumors were delineated within 20 minutes p.i.371 However, it became evident that there is high background radioactivity in the abdominal region (lung, liver, kidneys, spleen, and gallbladder) that hinders image interpretation.370
For PET studies, an 18F-labeled VIP analog has been evaluated by Jagoda et al.372 The lysine side chain of a VIP analog at position 20 was used for fluorination via N-succinimidyl-4-([18F]fluoromethyl) benzoate.58 The resulting radiopeptide [18F]dVIP was evaluated in vivo in a human breast xenograft mouse model, when revealing a two- to threefold higher [18F]FDG uptake in the tumor. However, the VIP receptor expression appeared to be rather low. The variety of radiolabeled VIP tracers all showed biologic properties comparable with the native VIP. In some studies, it was demonstrated that VIP receptor imaging could be possible. However, because of the endogenous expression on a variety of organs, the applicability of VIP receptor imaging is uncertain.
Radiolabeled Bombesin and Gastrin-Releasing Peptide Derivatives
Bombesin (BBN) is a 14 amino acid neuropeptide, originally found in frog skin, that displays high affinity for the gastrin-releasing peptide (GRP) receptor. BBN and the mammalian GRP with a sequence of 27 amino acids share the amidated C-terminal sequence Trp-Ala-Val-Gly-His-Leu-Met-NH2. Both peptides induce a variety of biologic responses in the central nervous system and in peripheral tissue, including the release of gastrointestinal hormones, smooth-muscle contraction, effects on blood pressure, and thermoregulation.373,374 Four bombesin receptor subtypes have been characterized,375 which are designated as the neuromedin B subtype (BB1), the GRP subtype (BB2), the orphan receptor subtype (BB3), and the BBN receptor subtype (BB4). From these, BB2 is overexpressed on a variety of tumors including prostate, breast, pancreas, and small-cell lung cancer,375 when it has been demonstrated that BB2 expression in primary prostatic invasive carcinoma was found in more than 90% of the tissue tested,376,377 whereas the expression in nonneoplastic prostatic tissue was low or completely absent. Interestingly, the endogenous expression of BB2 is limited to neuroendocrine cells in the pulmonary and gastrointestinal tracts, which would result in very low uptake of radiolabeled BBN analogs and a limited dose in normal tissue.378 Consequently, radiolabeled BBN analogs for nuclear medicine imaging and therapeutic applications have been proposed. These approaches are principally based on the C-terminal seven amino acid sequence as a lead structure for the development of a variety of radiolabeled derivatives.
The basic results for the structure–activity relationship of BBN and its derivatives have been demonstrated using radioiodinated Tyr4-BBN binding to rat brain membranes.379 Additional studies show that the amino acid sequence Trp-Ala-Val-Gly-His-Leu (BBN(8-13)) is involved in the interactions with the GRP receptor.380 The C-terminus–like derivative meta-iodophenyl-Des-Met14-BBN(7-13)NH2 (mIP-bombesin), showed promising properties in a competitive inhibition assay to target small-cell lung cancer and advantageous characteristics concerning cellular internalization and retention and significantly increased uptake in an ovarian tumor model when compared with [125I]Tyr4-BBN.378 Moreover, the radiosynthesis of the peptide using meta-[125I]iodophenyl-N-hydroxysuccinimide381 resulted in a tracer that showed reduced deiodination in vivo.
There are a large number of studies revealing the role of 99mTc/188Re-labeled BBN analogs. Most 99mTc-labeled bombesin derivatives are based on BBN(7-14)NH2. A variety of different chelating systems have been used, including dithiadiphosphine frameworks, N2S2, N3S, and N4 chelator systems and 99mTc(CO)3 complexes. Using a prelabeling procedure, [99mTc]P2S2-5-Ava-BBN(7-14)NH2 was synthesized.382 The IC50 value in vitro and tracer stability in vivo was satisfactory, providing important evidence that this water-soluble bifunctional chelator based on a P2S2 system is capable of stabilizing 99mTc under in vivo conditions. Blood clearance is rapid and tracer excretion occurs via both renal and hepatobiliary pathways. The major disadvantage of this approach is that the prelabeling procedure is time consuming and laborious.
One of the most prominent 99mTc chelating approaches is based on N3S chelators. Analogous to the DOTA derivatives,61 a limited series of 99mTc-labeled N3S-X-BBN(7-14)NH2 derivatives were synthesized by coupling the N3S chelator system (dimethyl)Gly-Ser-Cys-Gly via a spacer group to the peptide.59 Biodistribution data from nontumor–bearing CF-1 mice, along with data from the cell-binding studies, suggested that those conjugates where X = β-Ala, 5-Ava, or 8-Aoc are the most promising candidates for further in vivo evaluation. The preclinical studies with [99mTc]N3S-5-Ava-BBN(7-14)NH2 (99mTc-RP527) showed tumor/muscle ratios ranging from 6 to 25 at 4 hours p.i., depending on the tumor xenograft model used (PC-3, CF-PAC-1, AR42J). An initial study in patients showed 5 to 6 hours p.i. selective uptake in four of six breast carcinomas and one of four prostate carcinomas regardless of lesion size.383 Further biodistribution and dosimetry studies revealed low background in the thoracic region but intensive bowel activity, indicating that the identification of abdominal tumors may be problematic.384 The authors suggest use of this tracer for supradiaphragmatic region tumor imaging.
Besides N3S, the N2S2385 and N4-chelator systems53,386 were subjected to the formation of 99mTc(V) complexes. These complexes showed high affinity in vitro binding studies; some candidates based on the N4-chelator systems showed good tumor-to-background ratios allowing visualization of the tumor in vivo using a planar γ-camera. Interestingly, the agonist-based tracers do not translate to a higher tumor and target-organ localization in vivo compared to the antagonist-based [99mTc]Demobesin, as one might expect from the higher internalization rates of agonists. In addition, also 99mTc(I)-labeled bombesin analogs, such as [99mTc(CO)3Nα-histidinyl acetate]BBN(7-14)NH2, have been introduced.387,388 Despite high accumulation in the pancreas, [99mTc(CO)3Nα-histidinyl acetate]BBN(7-14)NH2 as well as [99mTc(I)Nα-PADA-AVA]BBN(7-14)NH2 showed low uptake in tumor in PC-3 tumor-bearing mice. It is speculated, that this may be caused by large difference in the blood supply between GRP receptor–positive acini cells in the pancreas and tumor cells. Low in vivo stability of the compounds may also add to the low tracer accumulation in the tumor.
99mTc(I)-labeled BBN analogs where diaminopropionic acid was used as a bidentate chelator have also been developed.59 For this series of 99mTc-labeled derivatives, the tracer uptake in the tumor and retention in xenografted human prostate cells in the murine models was superior to the 99mTc(I)-labeled BBN analogs introduced by La Bella et al.387,388 In addition, at 4 hours p.i., tracer uptake in tumor and pancreas were comparable, whereas radioactivity concentration in all other organs remained lower for [99mTc(P(CH2OH)3)(CO)3-Dpr-Ser-Ser-Ser]BBN(7-14)NH2, thus indicating that this peptide sequence is a determinant of the pharmacokinetics.
A large number of studies illustrate the role of radiometal-labeled BBN analogs, such as 111In-, 64Cu-, or 177Lu-labeled derivatives. Breeman et al.60 conjugated DTPA to the N-terminus of Pro1,Tyr4-BBN. This peptide had a nanomolar affinity to the BBN receptor and was internalized by BBN receptor–positive cells. Using CA20948 tumor-bearing rats, a very low accumulation of this tracer in the tumor was observed at 24 hours p.i. As expected, the predominant and much higher radioactivity concentration was found in kidneys and bladder. In a comparative study of DTPA and DOTA derivatives of Pro1,Tyr4-BBN and Lys3,Tyr4-BBN,389 the binding and internalization studies in vitro showed beneficial results for [111In]DTPA-Pro1,Tyr4-BBN followed by [111In]DTPA-Lys3,Tyr4-BBN and [111In]DOTA-Lys3,Tyr4-BBN. In accordance with the in vitro results, the tracer uptake in rat GRP receptor–positive pancreas ranged from 0.7%ID/g ([111In]DTPA-Lys3,Tyr4-BBN) to 1.3%ID/g ([111In]DOTA-Pro1,Tyr4-BBN) and was receptor-specific. However, although [111In]DOTA-Pro1,Tyr4-BBN showed the most suitable performance from this series of tracers, the corresponding DTPA analog was chosen for further clinical studies by the authors because of its more practical synthesis. Hoffman et al.61 studied a series of DOTA-X-BBN(7-14)NH2 derivatives where the chelator is conjugated via a spacer with variable hydrocarbon length (X = 0, β-Ala, 5-Ava, 8-Aoc, or 11-Aun) to the N-terminal end of the peptide. The in vitro competitive cell-binding assays demonstrated high specificity and affinity for the derivatives, when an optimal spacer length of three to eight carbon atoms was used. Importantly, the tracer uptake of 111In-labeled BBN derivatives in the GRP-receptor–expressing pancreas increased as a function of spacer length, reaching a maximum of approximately 27%ID/g at 1 hour p.i. in mice for the 8-Aoc spacer. However, in contrast to the in vitro data, no significant difference was found between the 8-Aoc spacer-bearing peptide and the peptide with the 11-Aun spacer. [111In]DOTA-8-Aoc-BBN(7-14)NH2 showed high tumor/blood and tumor/muscle ratios indicating that this molecule could be suitable as a lead for the further development of GRP tracers for human use.
As a consequence, this DOTA derivative was labeled with 177Lu62 and 64Cu390 to provide potential derivatives for therapy and for PET imaging studies.391 [177Lu]DOTA-8-Aoc-BBN(7-14)NH2 demonstrated very similar characteristics in vitro and in vivo when compared to the 111In-labeled derivative. [64Cu]DOTA-8-Aoc-BBN(7-14)NH2 also demonstrated high affinity for the GRP receptor in vitro and tumor uptake of approximately 4.2%ID/g at 4 hours p.i. in a murine tumor model. However, there was an unpredictably high concentration of radioactivity in normal tissue. Possibly, this could be explained by the differences in complex stability between the double-charged Cu(II) and the triple-charged Lu(III) and In(III). Moreover, it is well known that Cu(II) can be chelated by albumin and superoxide dismutase, resulting in blood activity and liver uptake.392,393 Interestingly, when the chelator is conjugated via the ∊-amino function of the lysine moiety to give [64Cu]DOTA-Lys3-BBN,394 the biodistribution and the tumor uptake in PC-3 tumor-bearing mice is comparable to other derivatives, such as [111In]DOTA-8-Aoc-BBN(7-14)NH2. The 64Cu-labeled polyethylene glycol conjugate with bombesin was synthesized,390 when biodistribution studies revealed that the PEG linker did not induce beneficial biokinetics or clearance properties in comparison with an alkyl linker. A comparison between 64Cu-DOTA-Aca-BBN(7-14) with 64Cu-DOTA-[Lys3]BBN did not show significant differences concerning their biodistribution, however, the authors assume that 64Cu-DOTA-[Lys3]BBN may have a better potential for clinical translation, because of the more practical synthesis.63
Using a PEG4 linker in DOTA-PEG4-BN(7-14) (DOTA-PESIN), 177Lu-labeling and the 67Ga-labeled analog were intensively studied in vitro and in vivo. The 177Lu-labeled peptide remained in the tumor 3 days p.i.395 When different chelators were used for the radiosyntheses of a series of BBN antagonists based on the sequence PEG4-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2, it became obvious that the chelators also influence affinity, antagonistic potency, and pharmacokinetics of the conjugates.396 Progress in chelator-based chemistry has facilitated further studies to assess tracer properties in vitro and in vivo by direct comparison of newly developed tracers to already known tracers.397–399 These comparative studies will likely significantly facilitate tracer development for PET and for therapy.
Only a limited number of publications have reported on 68Ga- or 18F-labeled bombesin peptides for PET imaging of GRP receptor expression.400,401 Schuhmacher et al.64 observed that the 68Ga-labeled derivative DOTA-PEG2-[D-Tyr6,β-Ala11,Thi13,Nle14]BN(6-14) amide (BZH3), had increased tumor retention and adequate clearance from receptor-negative tissue. The in vivo data of 68Ga-BZH3 indicate its potential for improved localization of GRP receptor–positive tumors.64 In a subsequent study, including 17 patients and a comparison with [18F]FDG, it is found that 68Ga-BZH3 may be helpful for diagnostic reasons in a subgroup of patients with gastrointestinal stromal tumors, especially those with negative [18F]FDG despite viable tumor tissue.402 More recently, preliminary PET kinetic data using 68Ga-BZH have been reported in patients with recurrent gliomas and compared to [18F]FDG PET data, when Ga-BZH seems to be helpful for the differentiation between low- and high-grade gliomas.403
As an approach toward 18F-labeled analogs, the use of di-tert-butyl silyl building blocks, direct nucleophilic labeling and labeling via activated esters have been applied.212,404,405 The direct 18F-labeling of an appropriate peptide precursor resulted in adequate and sufficient radiochemical yields and the in vivo PET imaging identified PC-3 tumor in nude mice.406 Based on this study, the 18F-labeled peptide 18F-BAY 86-4367, bearing three artificial amino acids within its peptide sequence, was characterized and displayed a subnanomolar receptor affinity. The favorable preclinical data showing specific and effective tumor targeting by 18F-BAY 86-4367 make this PET tracer a highly promising candidate to image prostate carcinoma.65
Recently, a variety of approaches have been reported on the multimeric and heterodimeric targeting of prostate cancer for imaging and therapeutic purposes, using bombesin derivatives.407–409 These studies report on the promising synergistic effect of a dual-receptor targeting of peptide heterodimers, such as RGD–bombesin conjugates, addressing tumors that express integrin or GRP receptors or that coexpress integrin and GRP receptors.
Neurotensin Analogs for Tumor Targeting
The neuropeptide neurotensin (NT), a tridecapeptide with the sequence pGlu-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Tyr-Ile-Leu-OH, was originally isolated from bovine hypothalamus.410 In mammalians, NT is localized in the central nervous system as well as in peripheral tissue, where it is mainly found in the gastrointestinal tract. NT acts as a neurotransmitter or neuromodulator of dopamine transmission and of anterior pituitary hormone secretion411,412as well as a local hormone exerting a paracrine and endocrine modulation of the digestive tract and of the cardiovascular system.412,413 NT binds with high affinity and low capacity to the neurotensin receptor subtype-1 (NTS1) and with low affinity and high capacity to the neurotensin receptor subtype-2 (NTS2).414,415 Both subtypes belong to the family of GPCRs and are internalized when the receptor–ligand complex is formed (Fig. 42.1). In addition, a membrane protein that binds NT with high affinity and is not G-protein–coupled has been identified.416 Because a variety of NETs (e.g., pancreatic adenocarcinoma, Ewing’s sarcoma, and meningioma) significantly overexpress neurotensin receptors, these receptors are molecular targets for radiolabeled neurotensin analogs for imaging applications or therapeutic interventions.417–419
Until now, the development of radiotracers was almost exclusively based on the C-terminal hexapeptide H-Arg-Arg-Pro-Tyr-Ile-Leu-OH (NT(8-13)) which is the minimal fragment for biologic activity.420,421 Chavatte et al.66reported on radioiodinated and 111In-DTPA–labeled NT(8-13) derivatives for tumor targeting and stated that labeling did not significantly influence binding affinity in vitro.
However, the major disadvantage of all these peptides based on NT(8-13) is the very low stability in vivo. The main proteolytic cleavage sites in the NT sequence are found to be between Arg8 and Arg9, Pro10 and Tyr11, and Tyr11and Ile12.422 Thus, a variety of peptides, where either one or more of these amide bonds were modified or corresponding amino acids were replaced by artificial amino acids, have been introduced. One approach to stabilization of the peptide sequence is replacement of the proteolytic unstable CO-NH function by a pseudo-peptide isosteric bond of the ψ(CH2NH) type, which is stable against hydrolysis. Chavatte et al.423 found that reduction of the bond between Arg8 and Arg9 and replacement of Arg8 by Lys resulted in a derivative with high binding affinity whereas replacement of other amide bonds caused a loss of receptor affinity. Following this approach, the biologic half-life in plasma could be improved from 1.5 (native NT) to 275 minutes (111In-DTPA-Lys8-ψ(CH2NH)-Arg9-NT(10-13)). Applying a similar approach for the synthesis of stabilized 99mTc-labeled NT(8-13) derivatives, the plasma half-life could be increased from 10 minutes for NT(8-13) to 180 minutes for (Nα-His)Ac-Lys8-ψ(CH2NH)-Arg9-NT(10-13) and 240 minutes for (Nα-His)Ac-Arg8-Arg9-NT(10-13) by retaining the binding properties of the native NT(8-13).422However, it was revealed that the main cleavage site of the stabilized peptides of this type was now between Tyr11 and Ile12. Consequently, further modification at this site was carried out to obtain NT derivatives stable enough for in vivo imaging applications. Therefore, the NT(8-13) sequence was further varied by Nα-methylation of Arg8 and replacement of Arg9 and Ile12 by Lys and tertiary leucin (Tle), respectively.424 (Nα-His)Ac was conjugated to the N-terminal end, allowing labeling via 99mTc(CO)3. These modifications resulted in a radiopeptide which remained intact in plasma for up to 24 hours at 37°C. The authors emphasize that stabilization of the amide bond between amino acids 11 and 12 is more important than stabilization of the Arg–Arg bond. The Tc-radiotracer was rapidly cleared from the blood, when highest uptake was found in kidneys and tumor at 1.5 hours after tracer injection in HT29-bearing nude mice. This approach was further optimized resulting in tracers with plasma half-lives of 20 days, and extended to the radiosynthesis of 188Re-labeled analogs with improved biokinetic properties, making such tracers promising candidates for clinical use.68,425
A comparable situation was found for (Nα-His)Ac-Lys8-ψ(CH2NH)-Arg9-Pro-Tyr-Tle-Leu-OH developed by Bruehlmeier et al.426 This 99mTc(CO)3-labeled peptide was used for an initial clinical study,427 including four patients with ductal pancreatic adenocarcinoma. The biologic half-lives of the intact tracer in blood were 35 and 230 minutes defined by a two-phase curve. However, only the patient with the highest receptor expression showed tracer accumulation in the tumor region, whereas immunohistochemistry of tumor slices revealed NT receptor expression for two of the four patients. Thus, further studies have to be carried out to fully elucidate the role and applicability of radiopeptides for imaging of NTS1 expression in vivo. As an extension of this approach, shikimic acid was introduced to the peptide and a triple stabilization was achieved by replacement of Tyr11 by 2,6-dimethyltyrosine (Dmt) to result in improved biokinetic properties of the peptide mimetic because of its increased hydrophilicity.428
An alternative approach to stabilize NT analogs is based on the introduction of artificial amino acids,67 such as [111In]DTPA-(Pip)Gly-Pro-(PipAm)Gly-Arg-Pro-Tyr-tBuGly-Leu-OH, showing high in vitro binding affinity and stability in serum, and receptor-mediated uptake in a murine tumor model. The radioactivity accumulation in receptor-negative tissue such as blood, spleen, pancreas, liver, muscle, and femur was low. Not surprisingly, highest uptake was found in kidneys. Anyway, this compound was suggested to be an excellent candidate for imaging and therapy of exocrine pancreatic cancer, which still remains to be demonstrated by in vivo studies.
In the field of 99mTc-labeled NT analogs, a series of N-terminal (Nα-His)Ac-linked peptides were studied by comparison of the Lys8-ψ(CH2NH)-Arg9 with Arg8-(NCH3)-Arg9 and the β3Arg8-Arg9 derivative, each with the Pro-Tyr-Tle-Leu sequence.425 The Arg8-(NCH3)-Arg9 derivative showed the best biodistribution in mice. Again, this result was further improved by using the Tyr to Dmt replacement, resulting in an increased half-life in plasma of the 99mTc-labeled peptide mimetic in vitro (28 days) and in vivo (blood, 1.5 hours), which makes this radiopeptide a promising candidate for clinical studies.68
Similar results were obtained when monocationic 99mTcO2+ tetraamine complexes attached at the N-terminus of NT analogs (including the Dab9-stabilized “Demostatin-6”) were studied.429 Very recently, the 99mTc-labeled Demostatin-6 was used in a first clinical study including 14 patients, when the tracer was well tolerated by patients and showed favorable pharmacokinetics; however, tumor targeting was limited to brain metastases.430
The development of 18F-labeled NT(8-13) analogues started with very little success.431 The 4-[18F]fluorobenzoyl-NT(8-13) as well as two analogs thereof which were stabilized either at the amide bond of Arg8-Arg9 by introduction of a ψ(CH2NH) group (mono stabilized), or at the amide bond of Arg8-Arg9 and by introducing a tertiary leucin (Tle) instead of Ile12 (double stabilized) showed only moderate uptake in the studied murine tumor models. Despite stabilization, high amounts of radioactive metabolites have been identified, resulting in insufficient uptake for in vivo imaging purposes.
Maschauer et al.69 have successfully applied an efficient strategy toward 18F-labeling with concomitant glycosylation for the synthesis of 18F-glycopeptides as imaging agents for PET. This strategy was combined with the development of a metabolically stable glycopeptide analog of NT(8–13), applying the replacements of Ile to Tle in position 12 of the amino acid sequence and replacing the Arg-Arg to NLys-Lys to introduce a peptoidic structure. In fact, the 18F-labeled glycopeptide demonstrated high receptor affinity in vitro, an outstanding in vivo stability, and small-animal PET studies successfully demonstrated specific visualization of NTS1 expression in vivo. The same metabolically stabilized amino acid sequence was applied to the conjugation of DOTA directly to the N-terminal end for labeling with Ga-68, resulting in a decreased NTS1 affinity; however, it was shown that this did not impair imaging of NTS1-positive tumors in HT29 xenografts.70 In addition, it has been shown recently that the introduction of a spacer between the DOTA chelator increased binding affinity, resulting in stabilized 68Ga-labeled peptide mimetics that were suitable for the detection of tumors with different sizes.432 Clearly, the kidney uptake of these radiopeptides has to be lowered, if the translation of this approach toward 177Lu-labeled peptides for radiotherapy could be considered.
In summary, because of the overexpression of NT receptors on a variety of tumors radiolabeled NT analogs for tumor imaging would be of great interest. To prevent proteolytic degradation, a variety of stabilized analogs have been introduced. However, further preclinical and clinical studies have to be carried out to fully evaluate the potential of radiopeptides derived from NT for radiotherapy and imaging applications.
SUMMARY AND EXPECTATIONS
Because the successful introduction of [18F]FDG and PET technology for imaging in oncology approximately 30 years ago, there has been an ongoing interest in radiopharmaceuticals for the noninvasive determination of molecular processes. A detailed characterization of the metabolic phenotype of malignancy is a major goal in personalized medicine. Radiopharmaceuticals supplying information about a variety of molecular processes are already available for clinical use. These include amino acid derivatives for the determination of amino acid transport and protein synthesis, nitroimidazole derivatives and a thiosemicarbazone for imaging hypoxia, choline and acetate derivatives for the determination of the lipid metabolism, annexin V derivatives for apoptosis imaging, nucleoside derivatives for imaging proliferation and reporter gene expression, and compounds targeting different molecular processes during tumor-induced angiogenesis. The most well known are the amino acids [11C]MET, [18F]FET, and [18F]FDOPA, the thymidine derivative [18F]FLT, the choline derivatives [18F]FCH and [18F]FECH, as well as the hypoxia marker [18F]FMISO. The use of the alternative hypoxia imaging agent [64Cu]ATSM is somewhat restricted because of the availability of 64Cu-copper.
To image apoptosis, and especially angiogenesis, a great variety of tracers have been introduced and are in different stages of development. The most prominent apoptosis imaging agents are based on annexin V of which the 99mTc-labeled compounds are the most intensively studied. Initial preclinical studies are ongoing for different caspase 3 inhibitors, but more studies are needed to assess the potential of these radiopharmaceuticals. Among the different molecular processes involved in angiogenesis, four target structures are in particular studied. Tracer development for these target structures include antagonists of the integrin αVβ3, inhibitors of MMPs, antibody fragments against the ED-B domain of fibronectin, and VEGF derivatives as well as VEGF-targeting antibodies. The development of potent radiolabeled probes to image MMP expression during tumor-induced angiogenesis is not yet successful. For the other target structures, different compounds have already been entered clinical trials with the most comprehensive studies carried out using [18F]Galacto-RGD and PET technology for the noninvasive determination of αVβ3 expression. However, further studies are needed.
Another area of radiopharmaceutical development is targeting receptors overexpressed on tumors. The most prominent receptor class is the SSTR family. A variety of tracers are used for diagnostic as well as therapeutic approaches including [68Ga]DOTA-TOC for PET imaging as well as [177Lu]DOTA-TATE and [90Y]DOTA-TOC for PRRT. In the past, it was postulated that internalizing agonists are the better compounds for this purpose. However, recently, it was demonstrated that SSTR antagonists like [111In]DOTA-BASS may perform better than the established agonists. Another well-known receptor-targeting radiopharmaceutical is [18F]FES for estrogen receptor imaging in breast cancer patients, which has been available for clinical investigations for more than two decades, although not FDA approved for routine clinical use.
A variety of other receptors are available as target structures for radiotracer development. This include the chemokine receptor CXCR4, the melanocortin-1 receptor, the neurokinin type 1 receptor, the CCK2/gastrin receptor, the neurotensin receptors, the bombesin receptors, and receptors for the VIP. Some have been studied for a long period (e.g., CCK-2, VIP, BBN), others have been evaluated more recently (e.g., NT, NK, α-MSH, CXCR4). Several approaches to increase metabolic stability are being studied including cyclization, peptide bond stabilization, and introduction of D-amino acids or artificial amino acids. The spectra of compounds range from very promising candidates in preclinical settings (e.g., [68Ga]CPCR4-2) to compounds which have been evaluated in clinical studies (e.g., [123I]VIP). It can be expected that additional molecular marker for the use with PET and SPECT technology and PRRT will become available for diagnosis and treatment in oncology.
REFERENCES
1. Hamacher K, Coenen HH, Stocklin G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986;27:235–238.
2. Del Fiore G, Schmitz P, Plenevaux A, et al. Fast routine production of l-[11C-methyl]methionine with Al2O3/KF in a disposable unit. J Labelled Comp Radiopharm. 1995;37:746–748.
3. Wester HJ, Herz M, Weber W, et al. Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-tyrosine for tumor imaging. J Nucl Med. 1999;40:205–212.
4. Hamacher K, Coenen HH. Efficient routine production of the 18F-labelled amino acid O-2-18F fluoroethyl-L-tyrosine. Appl Radiat Isot. 2002;57:853–856.
5. Fuchtner F, Zessin J, Mading P, et al. Aspects of 6-[18F]fluoro-L-DOPA preparation. Deuterochloroform as a substitute solvent for Freon 11. Nuklearmedizin. 2008;47:62–64.
6. Sorger D, Patt M, Kumar P, et al. [18F]Fluoroazomycinarabinofuranoside (18FAZA) and [18F]Fluoromisonidazole (18FMISO): A comparative study of their selective uptake in hypoxic cells and PET imaging in experimental rat tumors. Nucl Med Biol. 2003;30:317–326.
7. Dehdashti F, Grigsby PW, Lewis JS, et al. Assessing tumor hypoxia in cervical cancer by PET with 60Cu-labeled diacetyl-bis(N4-methylthiosemicarbazone). J Nucl Med. 2008;49:201–205.
8. Reischl G, Bieg C, Schmiedl O, et al. Highly efficient automated synthesis of [(11)C]choline for multi dose utilization. Appl Radiat Isot. 2004;60:835–838.
9. DeGrado TR, Baldwin SW, Wang S, et al. Synthesis and evaluation of (18)F-labeled choline analogs as oncologic PET tracers. J Nucl Med. 2001;42:1805–1814.
10. Hara T, Kosaka N, Kishi H. Development of (18)F-fluoroethylcholine for cancer imaging with PET: Synthesis, biochemistry, and prostate cancer imaging. J Nucl Med. 2002;43:187–199.
11. Ponde DE, Dence CS, Oyama N, et al. 18F-fluoroacetate: A potential acetate analog for prostate tumor imaging–in vivo evaluation of 18F-fluoroacetate versus 11C-acetate. J Nucl Med. 2007;48:420–428.
12. Machulla HJ, Blocher A, Kuntzsch M, et al. Simplified labeling approach for synthesizing 3′-deoxy-3′-[18F]fluorothymidine ([18F]FLT). J Radioanal Nucl Chem. 2000;243:843–846.
13. Brust P, Haubner R, Friedrich A, et al. Comparison of [18F]FHPG and [124/125I]FIAU for imaging herpes simplex virus type 1 thymidine kinase gene expression. Eur J Nucl Med. 2001;28:721–729.
14. Yaghoubi SS, Gambhir SS. PET imaging of herpes simplex virus type 1 thymidine kinase (HSV1-tk) or mutant HSV1-sr39tk reporter gene expression in mice and humans using [18F]FHBG. Nat Protoc. 2006;1:3069–3075.
15. Boersma HH, Liem IH, Kemerink GJ, et al. Comparison between human pharmacokinetics and imaging properties of two conjugation methods for 99mTc-annexin A5. Br J Radiol. 2003;76:553–560.
16. Kemerink GJ, Liu X, Kieffer D, et al. Safety, biodistribution, and dosimetry of 99mTc-HYNIC-annexin V, a novel human recombinant annexin V for human application. J Nucl Med. 2003;44:947–952.
17. Ke S, Wen X, Wu QP, et al. Imaging taxane-induced tumor apoptosis using PEGylated, 111In-labeled annexin V. J Nucl Med. 2004;45:108–115.
18. Murakami Y, Takamatsu H, Taki J, et al. 18F-labelled annexin V: A PET tracer for apoptosis imaging. Eur J Nucl Med Mol Imaging. 2004;31:469–474.
19. Li X, Link JM, Stekhova S, et al. Site-specific labeling of annexin V with F-18 for apoptosis imaging. Bioconjug Chem. 2008;19:1684–1688.
20. Haubner R, Kuhnast B, Mang C, et al. [18F]Galacto-RGD: Synthesis, radiolabeling, metabolic stability, and radiation dose estimates. Bioconjug Chem. 2004;15: 61–69.
21. Morrison MS, Ricketts SA, Barnett J, et al. Use of a novel Arg-Gly-Asp radioligand, 18F-AH111585, to determine changes in tumor vascularity after antitumor therapy. J Nucl Med. 2009;50:116–122.
22. Cho HJ, Lee JD, Park JY, et al. First in human evaluation of a newly developed PET tracer, 18F-RGD-K5 in patients with breast cancer: Comparison with 18F-FDG uptake pattern and microvessel density. J Nucl Med. 2009;50(suppl 2): 1910.
23. Mittra ES, Goris ML, Iagaru AH, et al. Pilot pharmacokinetic and dosimetric studies of (18)F-FPPRGD2: A PET radiopharmaceutical agent for imaging alpha(v)beta(3) integrin levels. Radiology. 2011;260:182–191.
24. Knetsch P, Petrik M, Rangger C, et al. Ga-68 labelled RGD peptide for monitoring angiogenesis. J Labelled Comp Radiopharm. 2009;52:S413.
25. Wagner S, Breyholz HJ, Faust A, et al. Molecular imaging of matrix metalloproteinases in vivo using small molecule inhibitors for SPECT and PET. Curr Med Chem. 2006;13:2819–2838.
26. Nagengast WB, de Vries EG, Hospers GA, et al. In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft. J Nucl Med. 2007; 48:1313–1319.
27. Jayson GC, Zweit J, Jackson A, et al. Molecular imaging and biological evaluation of HuMV833 anti-VEGF antibody: Implications for trial design of antiangiogenic antibodies. J Natl Cancer Inst. 2002;94:1484–1493.
28. Levashova Z, Backer M, Backer JM, et al. Direct site-specific labeling of the Cys-tag moiety in scVEGF with technetium 99m. Bioconjug Chem. 2008;19: 1049–1054.
29. Cai W, Chen K, Mohamedali KA, et al. PET of vascular endothelial growth factor receptor expression. J Nucl Med. 2006;47:2048–2056.
30. Tarli L, Balza E, Viti F, et al. A high-affinity human antibody that targets tumoral blood vessels. Blood. 1999;94:192–198.
31. Rossin R, Berndorff D, Friebe M, et al. Small-animal PET of tumor angiogenesis using a (76)Br-labeled human recombinant antibody fragment to the ED-B domain of fibronectin. J Nucl Med. 2007;48:1172–1179.
32. Bakker WH, Albert R, Bruns C, et al. [111In-DTPA-D-Phe1]-octreotide, a potential radiopharmaceutical for imaging of somatostatin receptor-positive tumors: Synthesis, radiolabeling and in vitro validation. Life Sci. 1991;49:1583– 1591.
33. Decristoforo C, Melendez-Alafort L, Sosabowski JK, et al. 99mTc-HYNIC-[Tyr3]-octreotide for imaging somatostatin-receptor-positive tumors: Preclinical evaluation and comparison with 111In-octreotide. J Nucl Med. 2000;41:1114–1119.
34. Breeman WA, de Jong M, de Blois E, et al. Radiolabelling DOTA-peptides with 68Ga. Eur J Nucl Med Mol Imaging. 2005;32:478–485.
35. Kayani I, Bomanji JB, Groves A, et al. Functional imaging of neuroendocrine tumors with combined PET/CT using 68Ga-DOTATATE (DOTA-DPhe1,Tyr3-octreotate) and 18F-FDG. Cancer. 2008;112:2447–2455.
36. Ambrosini V, Campana D, Bodei L, et al. 68Ga-DOTANOC PET/CT clinical impact in patients with neuroendocrine tumors. J Nucl Med. 2010;51:669–673.
37. Wild D, Fani M, Behe M, et al. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J Nucl Med. 2011;52:1412–1417.
38. Virgolini IJ, Gabriel M, von Guggenberg E, et al. Role of radiopharmaceuticals in the diagnosis and treatment of neuroendocrine tumours. Eur J Cancer. 2009;45(suppl 1):274–291.
39. Kiesewetter DO, Kilbourn MR, Landvatter SW, et al. Preparation of four fluorine- 18-labeled estrogens and their selective uptakes in target tissues of immature rats. J Nucl Med. 1984;25:1212–1221.
40. Nimmagadda S, Pullambhatla M, Stone K, et al. Molecular imaging of CXCR4 receptor expression in human cancer xenografts with [64Cu]AMD3100 positron emission tomography. Cancer Res. 2010;70:3935–3944.
41. Gourni E, Demmer O, Schottelius M, et al. PET of CXCR4 expression by a (68)Ga-labeled highly specific targeted contrast agent. J Nucl Med. 2011;52:1803–1810.
42. Demmer O, Dijkgraaf I, Schumacher U, et al. Design, synthesis, and functionalization of dimeric peptides targeting chemokine receptor CXCR4. J Med Chem. 2011;54:7648–7662.
43. Miao Y, Quinn TP. Alpha-melanocyte stimulating hormone peptide-targeted melanoma imaging. Front Biosci. 2007;12:4514–4524.
44. Quinn T, Zhang X, Miao Y. Targeted melanoma imaging and therapy with radiolabeled alpha-melanocyte stimulating hormone peptide analogues. G Ital Dermatol Venereol. 2010;145:245–258.
45. Wei L, Butcher C, Miao Y, et al. Synthesis and biologic evaluation of 64Cu-labeled rhenium-cyclized alpha-MSH peptide analog using a cross-bridged cyclam chelator. J Nucl Med. 2007;48:64–72.
46. Miao Y, Benwell K, Quinn TP. 99mTc- and 111In-labeled alpha-melanocyte-stimulating hormone peptides as imaging probes for primary and pulmonary metastatic melanoma detection. J Nucl Med. 2007;48:73–80.
47. Kneifel S, Cordier D, Good S, et al. Local targeting of malignant gliomas by the diffusible peptidic vector 1,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance p. Clin Cancer Res. 2006;12:3843–3850.
48. Cordier D, Forrer F, Bruchertseifer F, et al. Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8,Met(O2)11]-substance P: A pilot trial. Eur J Nucl Med Mol Imaging. 2010;37:1335–1344.
49. Mamelak AN, Rosenfeld S, Bucholz R, et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol. 2006;24:3644–3650.
50. Behr TM, Gotthardt M, Barth A, et al. Imaging tumors with peptide-based radioligands. Q J Nucl Med. 2001;45:189–200.
51. Good S, Walter MA, Waser B, et al. Macrocyclic chelator-coupled gastrin-based radiopharmaceuticals for targeting of gastrin receptor-expressing tumours. Eur J Nucl Med Mol Imaging. 2008;35:1868–1877.
52. von Guggenberg E, Sallegger W, Helbok A, et al. Cyclic minigastrin analogues for gastrin receptor scintigraphy with technetium-99m: Preclinical evaluation. J Med Chem. 2009;52:4786–4793.
53. Nock BA, Maina T, Behe M, et al. CCK-2/gastrin receptor-targeted tumor imaging with (99m)Tc-labeled minigastrin analogs. J Nucl Med. 2005;46:1727–1736.
54. Mather SJ, McKenzie AJ, Sosabowski JK, et al. Selection of radiolabeled gastrin analogs for peptide receptor-targeted radionuclide therapy. J Nucl Med. 2007; 48:615–622.
55. Reubi JC, Waser B, Schaer JC, et al. Unsulfated DTPA- and DOTA-CCK analogs as specific high-affinity ligands for CCK-B receptor-expressing human and rat tissues in vitro and in vivo. Eur J Nucl Med. 1998;25:481–490.
56. Laverman P, Behe M, Oyen WJ, et al. Two technetium-99m-labeled cholecystokinin-8 (CCK8) peptides for scintigraphic imaging of CCK receptors. Bioconjug Chem. 2004;15:561–568.
57. Virgolini I, Raderer M, Kurtaran A, et al. Vasoactive intestinal peptide-receptor imaging for the localization of intestinal adenocarcinomas and endocrine tumors. N Engl J Med. 1994;331:1116–1121.
58. Lang L, Eckelman WC. Labeling proteins at high specific activity using N-succinimidyl 4-[18F](fluoromethyl) benzoate. Appl Radiat Isot. 1997;48:169–173.
59. Smith CJ, Gali H, Sieckman GL, et al. Radiochemical investigations of (99m)Tc-N(3)S-X-BBN[7-14]NH(2): An in vitro/in vivo structure-activity relationship study where X = 0-, 3-, 5-, 8-, and 11-carbon tethering moieties. Bioconjug Chem. 2003;14:93–102.
60. Breeman WA, De Jong M, Bernard BF, et al. Pre-clinical evaluation of [(111)In-DTPA-Pro(1), Tyr(4)]bombesin, a new radioligand for bombesin-receptor scintigraphy. Int J Cancer. 1999;83:657–663.
61. Hoffman TJ, Gali H, Smith CJ, et al. Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J Nucl Med. 2003;44: 823–831.
62. Smith CJ, Gali H, Sieckman GL, et al. Radiochemical investigations of 177Lu-DOTA-8-Aoc-BBN[7-14]NH2: An in vitro/in vivo assessment of the targeting ability of this new radiopharmaceutical for PC-3 human prostate cancer cells. Nucl Med Biol. 2003;30:101–109.
63. Yang YS, Zhang X, Xiong Z, et al. Comparative in vitro and in vivo evaluation of two 64Cu-labeled bombesin analogs in a mouse model of human prostate adenocarcinoma. Nucl Med Biol. 2006;33:371–380.
64. Schuhmacher J, Zhang H, Doll J, et al. GRP receptor-targeted PET of a rat pancreas carcinoma xenograft in nude mice with a 68Ga-labeled bombesin(6-14) analog. J Nucl Med. 2005;46:691–699.
65. Honer M, Mu L, Stellfeld T, et al. 18F-labeled bombesin analog for specific and effective targeting of prostate tumors expressing gastrin-releasing peptide receptors. J Nucl Med. 2011;52:270–278.
66. Chavatte K, Mertens J, Terriere D, et al. Parameters ruling optimization of radiolabelling of polyamino polycarboxylated functionalized peptide derivatives: A case study report. Nucl Med Biol. 2001;28:745–749.
67. de Visser M, Janssen PJ, Srinivasan A, et al. Stabilised 111In-labelled DTPA- and DOTA-conjugated neurotensin analogues for imaging and therapy of exocrine pancreatic cancer. Eur J Nucl Med Mol Imaging. 2003;30:1134–1139.
68. Garcia-Garayoa E, Blauenstein P, Blanc A, et al. A stable neurotensin-based radiopharmaceutical for targeted imaging and therapy of neurotensin receptor-positive tumours. Eur J Nucl Med Mol Imaging. 2009;36:37–47.
69. Maschauer S, Einsiedel J, Haubner R, et al. Labeling and glycosylation of peptides using click chemistry: A general approach to (18)F-glycopeptides as effective imaging probes for positron emission tomography. Angew Chem Int Ed Engl. 2010;49:976–979.
70. Maschauer S, Einsiedel J, Hocke C, et al. Synthesis of a (68)Ga-labeled peptoid–peptide hybrid for imaging of neurotensin receptor expression in vivo. ACS Med Chem Lett. 2010;1:224–228.
71. Gregoire V, Haustermans K, Geets X, et al. PET-based treatment planning in radiotherapy: A new standard? J Nucl Med. 2007;48(suppl 1):68S–77S.
72. Plathow C, Weber WA. Tumor cell metabolism imaging. J Nucl Med. 2008; 49(suppl 2):43S–63S.
73. Yamamoto T, Seino Y, Fukumoto H, et al. Over-expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990;170: 223–230.
74. Arora KK, Fanciulli M, Pedersen PL. Glucose phosphorylation in tumor cells. Cloning, sequencing, and overexpression in active form of a full-length cDNA encoding a mitochondrial bindable form of hexokinase. J Biol Chem. 1990;265: 6481–6488.
75. Maschauer S, Prante O, Hoffmann M, et al. Characterization of 18F-FDG uptake in human endothelial cells in vitro. J Nucl Med. 2004;45:455–460.
76. Aloj L, Caraco C, Jagoda E, et al. Glut-1 and hexokinase expression: Relationship with 2-fluoro-2-deoxy-D-glucose uptake in A431 and T47D cells in culture. Cancer Res. 1999;59:4709–4714.
77. Ko BH, Paik JY, Jung KH, et al. 17beta-estradiol augments 18F-FDG uptake and glycolysis of T47D breast cancer cells via membrane-initiated rapid PI3K-Akt activation. J Nucl Med. 2010;51:1740–1747.
78. Treglia G, Mattoli MV, Leccisotti L, et al. Usefulness of whole-body fluorine-18-fluorodeoxyglucose positron emission tomography in patients with large-vessel vasculitis: A systematic review. Clin Rheumatol. 2011;30:1265–1275.
79. Ogawa M, Nakamura S, Saito Y, et al. What can be seen by 18F-FDG PET in atherosclerosis imaging? The effect of foam cell formation on 18F-FDG uptake to macrophages in vitro. J Nucl Med. 2012;53:55–58.
80. Deichen JT, Prante O, Gack M, et al. Uptake of [18F]fluorodeoxyglucose in human monocyte-macrophages in vitro. Eur J Nucl Med Mol Imaging. 2003;30: 267–273.
81. Kubota R, Yamada S, Kubota K, et al. Intratumoral distribution of fluorine-18-fluorodeoxyglucose in vivo: High accumulation in macrophages and granulation tissues studied by microautoradiography. J Nucl Med. 1992;33:1972–1980.
82. Clavo AC, Brown RS, Wahl RL. Fluorodeoxyglucose uptake in human cancer cell lines is increased by hypoxia. J Nucl Med. 1995;36:1625–1632.
83. Folco EJ, Sheikine Y, Rocha VZ, et al. Hypoxia but not inflammation augments glucose uptake in human macrophages: Implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-D-glucose positron emission tomography. J Am Coll Cardiol. 2011;58:603–614.
84. Buck AK, Schirrmeister H, Mattfeldt T, et al. Biological characterisation of breast cancer by means of PET. Eur J Nucl Med Mol Imaging. 2004;31:S80–S87.
85. Paik JY, Lee KH, Choe YS, et al. Augmented 18F-FDG uptake in activated monocytes occurs during the priming process and involves tyrosine kinases and protein kinase C. J Nucl Med. 2004;45:124–128.
86. Haberkorn U, Altmann A, Kamencic H, et al. Glucose transport and apoptosis after gene therapy with HSV thymidine kinase. Eur J Nucl Med. 2001;28:1690–1696.
87. Paik JY, Lee KH, Ko BH, et al. Nitric oxide stimulates 18F-FDG uptake in human endothelial cells through increased hexokinase activity and GLUT1 expression. J Nucl Med. 2005;46:365–370.
88. Ido T, Wan CN, Fowler JS, et al. Fluorination with F2-2. A convenient synthesis of 2-FDG. J Org Chem. 1977;42:2341–2342.
89. Bida GT, Satyamurthy N, Barrio JR. The synthesis of 2-[F-18]fluoro-2-deoxy-D-glucose using glycals: A reexamination. J Nucl Med. 1984;25:1327–1334.
90. Yu S. Review of F-18-FDG synthesis and quality control. Biomed Imaging Interv J. 2006;2:e57.
91. Hawkins RA, Huang SC, Barrio JR, et al. Estimation of local cerebral protein synthesis rates with L-[1-11C]leucine and PET: Methods, model, and results in animals and humans. J Cereb Blood Flow Metab. 1989;9:446–460.
92. Kubota K, Yamada K, Fukada H, et al. Tumor detection with carbon-11-labelled amino acids. Eur J Nucl Med. 1984;9:136–140.
93. Pruim J, Willemsen AT, Molenaar WM, et al. Brain tumors: L-[1-C-11]tyrosine PET for visualization and quantification of protein synthesis rate. Radiology. 1995;197:221–226.
94. McConathy J, Goodman MM. Non-natural amino acids for tumor imaging using positron emission tomography and single photon emission computed tomography. Cancer Metastasis Rev. 2008;27:555–573.
95. Langen KJ, Hamacher K, Weckesser M, et al. O-(2-[18F]fluoroethyl)-L-tyrosine: Uptake mechanisms and clinical applications. Nucl Med Biol. 2006;33:287–294.
96. Jager PL, Vaalburg W, Pruim J, et al. Radiolabeled amino acids: Basic aspects and clinical applications in oncology. J Nucl Med. 2001;42:432–445.
97. Shotwell MA, Jayme DW, Kilberg MS, et al. Neutral amino acid transport systems in Chinese hamster ovary cells. J Biol Chem. 1981;256:5422–5427.
98. Segawa H, Fukasawa Y, Miyamoto K-I, et al. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J Biol Chem. 1999;274:19745–19751.
99. Deves R, Boyd CA. Transporters for cationic amino acids in animal cells: Discovery, structure, and function. Physiol Rev. 1998;78:487–545.
100. Kanai Y, Segawa H, Miyamoto K, et al. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J Biol Chem. 1998;273:23629–23632.
101. Pineda M, Fernandez E, Torrents D, et al. Identification of a membrane protein, LAT-2, that Co-expresses with 4F2 heavy chain, an L-type amino acid transport activity with broad specificity for small and large zwitterionic amino acids. J Biol Chem. 1999;274:19738–19744.
102. Babu E, Kanai Y, Chairoungdua A, et al. Identification of a novel system L amino acid transporter structurally distinct from heterodimeric amino acid transporters. J Biol Chem. 2003;278:43838–43845.
103. Bodoy S, Martin L, Zorzano A, et al. Identification of LAT4, a novel amino acid transporter with system L activity. J Biol Chem. 2005;280:12002–12011.
104. Langen KJ, Jarosch M, Muhlensiepen H, et al. Comparison of fluorotyrosines and methionine uptake in F98 rat gliomas. Nucl Med Biol. 2003;30:501–508.
105. Stöber B, Tanase U, Herz M, et al. Differentiation of tumour and inflammation: Characterisation of [methyl-3H]methionine (MET) and O-(2-[18F]fluoroethyl)-L-tyrosine (FET) uptake in human tumour and inflammatory cells. Eur J Nucl Med Mol Imaging. 2006;33:932–939.
106. Pauleit D, Stoffels G, Bachofner A, et al. Comparison of (18)F-FET and (18)F-FDG PET in brain tumors. Nucl Med Biol. 2009;36:779–787.
107. Pauleit D, Stoffels G, Schaden W, et al. PET with O-(2-[18F]fluoroethyl)-L-tyrosine in peripheral tumors: First clinical results. J Nucl Med. 2005;46:411–416.
108. Pauleit D, Floeth F, Hamacher K, et al. O-(2- F-18 fluoroethyl)-L-tyrosine PET combined with MRI improves the diagnostic assessment of cerebral gliomas. Brain. 2005;128:678–687.
109. Floeth FW, Pauleit D, Wittsack HJ, et al. Multimodal metabolic imaging of cerebral gliomas: Positron emission tomography with F-18 fluoroethyl-L-tyrosine and magnetic resonance spectroscopy. J Neurosurg. 2005;102:318–327.
110. Pöpperl G, Goldbrunner R, Gildehaus FJ, et al. O-(2-[18F]fluoroethyl)-L-tyrosine PET for monitoring the effects of convection-enhanced delivery of paclitaxel in patients with recurrent glioblastoma. Eur J Nucl Med Mol Imaging. 2005;32:1018–1025.
111. Stadlbauer A, Prante O, Nimsky C, et al. Metabolic imaging of cerebral gliomas: Spatial correlation of changes in O-(2-18F-fluoroethyl)-L-tyrosine PET and proton magnetic resonance spectroscopic imaging. J Nucl Med. 2008;49:721–729.
112. Piroth MD, Pinkawa M, Holy R, et al. Integrated boost IMRT with FET-PET-adapted local dose escalation in glioblastomas : Results of a prospective phase II study. Strahlenther Onkol. 2012;188(4):334–339.
113. Salber D, Stoffels G, Oros-Peusquens AM, et al. Comparison of O-(2-18F-fluoroethyl)-L-tyrosine and L-3H-methionine uptake in cerebral hematomas. J Nucl Med. 2010;51:790–797.
114. Barrio JR, Huang SC, Phelps ME. Biological imaging and the molecular basis of dopaminergic diseases. Biochem Pharmacol. 1997;54:341–348.
115. Stout DB, Huang SC, Melega WP, et al. Effects of large neutral amino acid concentrations on 6-[F-18]Fluoro-L-DOPA kinetics. J Cereb Blood Flow Metab. 1998;18: 43–51.
116. Sundin A, Garske U, Orlefors H. Nuclear imaging of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:69–85.
117. Haug A, Auernhammer CJ, Wangler B, et al. Intraindividual comparison of 68Ga-DOTA-TATE and 18F-DOPA PET in patients with well-differentiated metastatic neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2009;36:765–770.
118. Padhani AR, Krohn KA, Lewis JS, et al. Imaging oxygenation of human tumours. Eur Radiol. 2007;17:861–872.
119. Piert M, Machulla HJ, Picchio M, et al. Hypoxia-specific tumor imaging with 18F-fluoroazomycin arabinoside. J Nucl Med. 2005;46:106–113.
120. Vavere AL, Lewis JS. Cu-ATSM: A radiopharmaceutical for the PET imaging of hypoxia. Dalton Trans. 2007:4893–4902.
121. Holland JP, Barnard PJ, Collison D, et al. Spectroelectrochemical and computational studies on the mechanism of hypoxia selectivity of copper radiopharmaceuticals. Chemistry. 2008;14:5890–5907.
122. Bourgeois M, Rajerison H, Guerard F, et al. Contribution of [64Cu]-ATSM PET in molecular imaging of tumour hypoxia compared to classical [18F]-MISO–a selected review. Nucl Med Rev Cent East Eur. 2011;14:90–95.
123. Bowen SR, van der Kogel AJ, Nordsmark M, et al. Characterization of positron emission tomography hypoxia tracer uptake and tissue oxygenation via electrochemical modeling. Nucl Med Biol. 2011;38:771–780.
124. Matsumoto K, Szajek L, Krishna MC, et al. The influence of tumor oxygenation on hypoxia imaging in murine squamous cell carcinoma using [64Cu]Cu-ATSM or [18F]Fluoromisonidazole positron emission tomography. Int J Oncol. 2007;30: 873–881.
125. O’Donoghue JA, Zanzonico P, Pugachev A, et al. Assessment of regional tumor hypoxia using 18F-fluoromisonidazole and 64Cu(II)-diacetyl-bis(N4-methylthiosemicarbazone) positron emission tomography: Comparative study featuring microPET imaging, Po2 probe measurement, autoradiography, and fluorescent microscopy in the R3327-AT and FaDu rat tumor models. Int J Radiat Oncol Biol Phys. 2005;61:1493–1502.
126. Emonds KM, Swinnen JV, Mortelmans L, et al. Molecular imaging of prostate cancer. Methods. 2009;48:193–199.
127. Moerlein SM, Gaehle GG, Welch MJ. Robotic preparation of Sodium Acetate C 11 Injection for use in clinical PET. Nucl Med Biol. 2002;29:613–621.
128. de Certaines JD, Larsen VA, Podo F, et al. In vivo 31P MRS of experimental tumours. NMR Biomed. 1993;6:345–365.
129. Shindou H, Shimizu T. Acyl-CoA: Lysophospholipid acyltransferases. J Biol Chem. 2009;284:1–5.
130. Deves R, Krupka RM. The binding and translocation steps in transport as related to substrate structure. A study of the choline carrier of erythrocytes. Biochim Biophys Acta. 1979;557:469–485.
131. Clary GL, Tsai CF, Guynn RW. Substrate specificity of choline kinase. Arch Biochem Biophys. 1987;254:214–221.
132. Patel SS, Walt DR. Substrate specificity of acetyl coenzyme A synthetase. J Biol Chem. 1987;262:7132–7134.
133. Zhang J-P, Zhang Y-J, Xu J-Y, et al. Radiation dosimetry estimates of [18F]-fluoroacetate based on biodistribution data of rats. Appl Radiat Isot. 2012;70: 332–335.
134. Nishii R, Tong W, Wendt R, et al. Pharmacokinetics, metabolism, biodistribution, radiation dosimetry, and toxicology of (18)F-Fluoroacetate ((18)F-FACE) in non-human primates. Mol Imaging Biol. 2012;14(2):213–224.
135. Buck AK, Herrmann K, Shen C, et al. Molecular imaging of proliferation in vivo: Positron emission tomography with [18F]fluorothymidine. Methods. 2009;48: 205–215.
136. Shields AF, Mankoff D, Graham MM, et al. Analysis of 2-carbon-11-thymidine blood metabolites in PET imaging. J Nucl Med. 1996;37:290–296.
137. Barwick T, Bencherif B, Mountz JM, et al. Molecular PET and PET/CT imaging of tumour cell proliferation using F-18 fluoro-L-thymidine: A comprehensive evaluation. Nucl Med Commun. 2009;30:908–917.
138. Grierson JR, Shields AF. Radiosynthesis of 3′-deoxy-3′-[(18)F]fluorothymidine: [(18)F]FLT for imaging of cellular proliferation in vivo. Nucl Med Biol. 2000;27: 143–156.
139. Martin SJ, Eisenbarth JA, Wagner-Utermann U, et al. A new precursor for the radiosynthesis of [18F]FLT. Nucl Med Biol. 2002;29:263–273.
140. Shields AF, Briston DA, Chandupatla S, et al. A simplified analysis of [18F]3′-deoxy-3′-fluorothymidine metabolism and retention. Eur J Nucl Med Mol Imaging. 2005;32:1269–1275.
141. Soloviev D, Lewis D, Honess D, et al. [18F]FLT: An imaging biomarker of tumour proliferation for assessment of tumour response to treatment. Euro J Cancer. 2012;48:416–424.
142. Contractor KB, Kenny LM, Stebbing J, et al. [18F]-3′deoxy-3′-fluorothymidine positron emission tomography and breast cancer response to docetaxel. Clin Cancer Res. 2011;17:7664–7672.
143. Lee SJ, Kim SY, Chung JH, et al. Induction of thymidine kinase 1 after 5-fluorouracil as a mechanism for 3′-deoxy-3′-[18F]fluorothymidine flare. Biochem Pharmacol. 2010;80:1528–1536.
144. Francis DL, Visvikis D, Costa DC, et al. Potential impact of [18F]3′-deoxy-3′-fluorothymidine versus [18F]fluoro-2-deoxy-D-glucose in positron emission tomography for colorectal cancer. Eur J Nucl Med Mol Imaging. 2003;30:988–994.
145. Min JJ, GAmbhir SS. Molecular imaging of PET reporter gene expression. In: Semmler W, Schwaiger M, eds. Molecular Imaging II. Handbook of Experimental Pharmacology. Berlin, Heidelberg: Springer; 2008:277–303.
146. Jacobs A, Braunlich I, Graf R, et al. Quantitative kinetics of [124I]FIAU in cat and man. J Nucl Med. 2001;42:467–475.
147. Blasberg RG, Tjuvajev JG. Molecular-genetic imaging: Current and future perspectives. J Clin Invest. 2003;111:1620–1629.
148. Cho JY. A transporter gene (sodium iodide symporter) for dual purposes in gene therapy: Imaging and therapy. Curr Gene Ther. 2002;2:393–402.
149. Verwijnen SM, Sillevis Smith PA, Hoeben RC, et al. Molecular imaging and treatment of malignant gliomas following adenoviral transfer of the herpes simplex virus-thymidine kinase gene and the somatostatin receptor subtype 2 gene. Cancer Biother Radiopharm.2004;19:111–120.
150. Anton M, Wagner B, Haubner R, et al. Use of the norepinephrine transporter as a reporter gene for non-invasive imaging of genetically modified cells. J Gene Med. 2004;6:119–126.
151. Alauddin MM, Gelovani JG. Radiolabeled nucleoside analogues for PET imaging of HSV1-tk gene expression. Curr Top Med Chem. 2010;10:1617–1632.
152. Haubner R, Avril N, Hantzopoulos PA, et al. In vivo imaging of herpes simplex virus type 1 thymidine kinase gene expression: Early kinetics of radiolabelled FIAU. Eur J Nucl Med. 2000;27:283–291.
153. Kang KW, Min JJ, Chen X, et al. Comparison of [14C]FMAU, [3H]FEAU, [14C]FIAU, and [3H]PCV for monitoring reporter gene expression of wild type and mutant herpes simplex virus type 1 thymidine kinase in cell culture. Mol Imaging Biol. 2005;7:296–303.
154. Iyer M, Barrio JR, Namavari M, et al. 8-[18F]Fluoropenciclovir: An improved reporter probe for imaging HSV1-tk reporter gene expression in vivo using PET. J Nucl Med. 2001;42:96–105.
155. Yaghoubi S, Barrio JR, Dahlbom M, et al. Human pharmacokinetic and dosimetry studies of [(18)F]FHBG: A reporter probe for imaging herpes simplex virus type-1 thymidine kinase reporter gene expression. J Nucl Med. 2001;42:1225–1234.
156. de Vries EF, van Dillen IJ, van Waarde A, et al. Evaluation of [18F]FHPG as PET tracer for HSVtk gene expression. Nucl Med Biol. 2003;30:651–660.
157. Tjuvajev JG, Avril N, Oku T, et al. Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Res. 1998;58: 4333–4341.
158. Vaidyanathan G, Zalutsky MR. Preparation of 5-[131I]iodo- and 5-[211At]astato-1-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) uracil by a halodestannylation reaction. Nucl Med Biol. 1998;25:487–496.
159. Tjuvajev JG, Doubrovin M, Akhurst T, et al. Comparison of radiolabeled nucleoside probes (FIAU, FHBG, and FHPG) for PET imaging of HSV1-tk gene expression. J Nucl Med. 2002;43:1072–1083.
160. Cai H, Li Z, Conti PS. The improved syntheses of 5-substituted 2′-[18F]fluoro-2′-deoxy-arabinofuranosyluracil derivatives ([18F]FAU, [18F]FEAU, [18F]FFAU, [18F]FCAU, [18F]FBAU and [18F]FIAU) using a multistep one-pot strategy. Nucl Med Biol. 2011;38:659–666.
161. Blankenberg FG, Norfray JF. Multimodality molecular imaging of apoptosis in oncology. AJR Am J Roentgenol. 2011;197:308–317.
162. Huerta S, Goulet EJ, Huerta-Yepez S, et al. Screening and detection of apoptosis. J Surg Res. 2007;139:143–156.
163. Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–988.
164. Andree HA, Reutelingsperger CP, Hauptmann R, et al. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J Biol Chem. 1990;265:4923–4928.
165. Kapty J, Murray D, Mercer J. Radiotracers for noninvasive molecular imaging of tumor cell death. Cancer Biother Radiopharm. 2010;25:615–628.
166. Ohtsuki K, Akashi K, Aoka Y, et al. Technetium-99m HYNIC-annexin V: A potential radiopharmaceutical for the in-vivo detection of apoptosis. Eur J Nucl Med. 1999;26:1251–1258.
167. Van den Brande JM, Koehler TC, Zelinkova Z, et al. Prediction of antitumour necrosis factor clinical efficacy by real-time visualisation of apoptosis in patients with Crohn’s disease. Gut. 2007;56:509–517.
168. Tait JF, Brown DS, Gibson DF, et al. Development and characterization of annexin V mutants with endogenous chelation sites for (99m)Tc. Bioconjug Chem. 2000;11:918–925.
169. Tait JF, Smith C, Blankenberg FG. Structural requirements for in vivo detection of cell death with 99mTc-annexin V. J Nucl Med. 2005;46:807–815.
170. Bauwens M, De Saint-Hubert M, Devos E, et al. Site-specific 68Ga-labeled Annexin A5 as a PET imaging agent for apoptosis. Nucl Med Biol. 2011;38:381–392.
171. Chen K, Chen X. Positron emission tomography imaging of cancer biology: Current status and future prospects. Semin Oncol. 2011;38:70–86.
172. Kopka K, Faust A, Keul P, et al. 5-pyrrolidinylsulfonyl isatins as a potential tool for the molecular imaging of caspases in apoptosis. J Med Chem. 2006;49:6704–6715.
173. Chu W, Zhang J, Zeng C, et al. N-benzylisatin sulfonamide analogues as potent caspase-3 inhibitors: Synthesis, in vitro activity, and molecular modeling studies. J Med Chem. 2005;48:7637–7647.
174. Podichetty AK, Wagner S, Schroer S, et al. Fluorinated isatin derivatives. Part 2. New N-substituted 5-pyrrolidinylsulfonyl isatins as potential tools for molecular imaging of caspases in apoptosis. J Med Chem. 2009;52:3484–3495.
175. Nguyen QD, Smith G, Glaser M, et al. Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-3/7 specific [18F]-labeled isatin sulfonamide. Proc Natl Acad Sci U S A. 2009;106:16375–16380.
176. Ellis LM, Liu W, Fan F, et al. Synopsis of angiogenesis inhibitors in oncology. Oncology. 2002;16:14–22.
177. Kuwano M, Fukushi J, Okamoto M, et al. Angiogenesis factors. Intern Med. 2001;40:565–572.
178. Creamer D, Sullivan D, Bicknell R, et al. Angiogenesis in psoriasis. Angiogenesis. 2002;5:231–236.
179. Bishop GG, McPherson JA, Sanders JM, et al. Selective alpha(v)beta(3)-receptor blockade reduces macrophage infiltration and restenosis after balloon angioplasty in the atherosclerotic rabbit. Circulation. 2001;103:1906–1911.
180. Storgard CM, Stupack DG, Jonczyk A, et al. Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist [see comments]. J Clin Invest. 1999;103:47–54.
181. Chavakis E, Riecke B, Lin J, et al. Kinetics of integrin expression in the mouse model of proliferative retinopathy and success of secondary intervention with cyclic RGD peptides. Diabetologia. 2002;45:262–267.
182. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18.
183. Loges S, Roncal C, Carmeliet P. Development of targeted angiogenic medicine. J Thromb Haemost. 2009;7:21–33.
184. Dumont RA, Hildebrandt I, Su H, et al. Noninvasive imaging of alphaVbeta3 function as a predictor of the antimigratory and antiproliferative effects of dasatinib. Cancer Res. 2009;69:3173–3179.
185. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000; 407:249–257.
186. Hagedorn M, Bikfalvi A. Target molecules for anti-angiogenic therapy: From basic research to clinical trials. Crit Rev Oncol Hematol. 2000;34:89–110.
187. Vihinen P, Kahari VM. Matrix metalloproteinases in cancer: Prognostic markers and therapeutic targets. Int J Cancer. 2002;99:157–166.
188. Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267–285.
189. Hynes RO, Bader BL, Hodivala-Dilke K. Integrins in vascular development. Braz J Med Biol Res. 1999;32:501–510.
190. Eliceiri BP, Cheresh DA. Role of alpha v integrins during angiogenesis. Cancer J Sci Am. 2000;6:S245–S249.
191. Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–617.
192. Brooks PC, Montgomery AM, Rosenfeld M, et al. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–1164.
193. Brooks PC, Stromblad S, Klemke R, et al. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin [see comments]. J Clin Invest. 1995;96:1815–1822.
194. Hammes HP, Brownlee M, Jonczyk A, et al. Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat Med. 1996;2:529–533.
195. Taga T, Suzuki A, Gonzalez-Gomez I, et al. alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer. 2002;98:690–697.
196. Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–921.
197. Rosen L. Antiangiogenic strategies and agents in clinical trials. Oncologist. 2000;1:20–27.
198. Gasparini G, Longo R, Toi M, et al. Angiogenic inhibitors: A new therapeutic strategy in oncology. Nat Clin Pract Oncol. 2005;2:562–577.
199. Dredge K, Dalgleish AG, Marriott JB. Recent developments in antiangiogenic therapy. Expert Opin Biol Ther. 2002;2:953–966.
200. Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100.
201. Ruoslahti E. Specialization of tumour vasculature. Nat Rev Cancer. 2002;2: 83–90.
202. Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497.
203. Aumailley M, Gurrath M, Muller G, et al. Arg-Gly-Asp constrained within cyclic pentapeptides. Strong and selective inhibitors of cell adhesion to vitronectin and laminin fragment P1. FEBS Lett. 1991;291:50–54.
204. Haubner RH, Wester HJ, Weber WA, et al. Radiotracer-based strategies to image angiogenesis. Q J Nucl Med. 2003;47:189–199.
205. Haubner R, Wester HJ, Weber WA, et al. Noninvasive imaging of alpha(v)beta3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61:1781–1785.
206. Haubner R, Weber WA, Beer AJ, et al. Noninvasive visualization of the activated alphavbeta3 integrin in cancer patients by positron emission tomography and [18F]Galacto-RGD. PLoS Med. 2005;2:e70.
207. Beer AJ, Haubner R, Sarbia M, et al. Positron emission tomography using [18F]Galacto-RGD identifies the level of integrin alpha(v)beta3 expression in man. Clin Cancer Res. 2006;12:3942–3949.
208. Beer AJ, Haubner R, Wolf I, et al. PET-based human dosimetry of 18F-galacto-RGD, a new radiotracer for imaging alpha v beta3 expression. J Nucl Med. 2006;47:763–769.
209. Beer AJ, Grosu AL, Carlsen J, et al. [18F]Galacto-RGD positron emission tomography for imaging of {alpha}v{beta}3 expression on the neovasculature in patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6610–6616.
210. Poethko T, Schottelius M, Thumshirn G, et al. Chemoselective pre-conjugate radiohalogenation of unprotected mono- and multimeric peptides via oxime formation. Radiochimica Acta. 2004;92:317–327.
211. Poethko T, Schottelius M, Thumshirn G, et al. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and octreotide analogs. J Nucl Med. 2004;45:892–902.
212. Schirrmacher E, Wangler B, Cypryk M, et al. Synthesis of p-(di-tert-butyl[(18)F]fluorosilyl)benzaldehyde ([(18)F]SiFA-A) with high specific activity by isotopic exchange: A convenient labeling synthon for the (18)F-labeling of N-amino-oxy derivatized peptides. Bioconjug Chem. 2007;18:2085–2089.
213. Lee YS, Jeong JM, Kim HW, et al. An improved method of 18F peptide labeling: Hydrazone formation with HYNIC-conjugated c(RGDyK). Nucl Med Biol. 2006; 33:677–683.
214. Glaser M, Morrison M, Solbakken M, et al. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjug Chem. 2008;19:951–957.
215. Prante O, Einsiedel J, Haubner R, et al. 3,4,6-Tri-O-acetyl-2-deoxy-2-[18F]fluoroglucopyranosyl phenylthiosulfonate: A thiol-reactive agent for the chemoselective 18F-glycosylation of peptides. Bioconjug Chem. 2007;18:254–262.
216. Cai W, Zhang X, Wu Y, et al. A thiol-reactive 18F-labeling agent, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide, and synthesis of RGD peptide-based tracer for PET imaging of alpha v beta 3 integrin expression. J Nucl Med. 2006;47: 1172–1180.
217. Glaser M, Solbakken M, Turton DR, et al. Methods for 18F-labeling of RGD peptides: Comparison of aminooxy [18F]fluorobenzaldehyde condensation with ‘click labeling’ using 2-[18F]fluoroethylazide, and S-alkylation with [18F]fluoropropanethiol. Amino Acids.2009;37:717–724.
218. Kolb H, Walsh J, Liang Q, et al. 18F-RGD-K5: A cyclic triazole-bearing RGD peptide for imaging integrin avb3 expression in vivo. J Nucl Med. 2009;50(suppl 2): 329.
219. Chen X, Park R, Tohme M, et al. MicroPET and autoradiographic imaging of breast cancer alpha v-integrin expression using 18F- and 64Cu-labeled RGD peptide. Bioconjug Chem. 2004;15:41–49.
220. Decristoforo C, Hernandez Gonzalez I, Carlsen J, et al. (68)Ga- and (111)In-labelled DOTA-RGD peptides for imaging of alphavbeta3 integrin expression. Eur J Nucl Med Mol Imaging. 2008;35(8):1507–1515.
221. Jeong JM, Hong MK, Chang YS, et al. Preparation of a promising angiogenesis PET imaging agent: 68Ga-labeled c(RGDyK)-isothiocyanatobenzyl-1,4,7-triazacyclononane-1,4,7-triacetic acid and feasibility studies in mice. J Nucl Med. 2008;49: 830–836.
222. Janssen ML, Oyen WJ, Dijkgraaf I, et al. Tumor targeting with radiolabeled alpha(v)beta(3) integrin binding peptides in a nude mouse model. Cancer Res. 2002;62:6146–6151.
223. Janssen M, Oyen WJ, Massuger LF, et al. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm. 2002;17:641–646.
224. Thumshirn G, Hersel U, Goodman SL, et al. Multimeric cyclic RGD peptides as potential tools for tumor targeting: Solid-phase peptide synthesis and chemoselective oxime ligation. Chemistry. 2003;9:2717–2725.
225. Chen X, Tohme M, Park R, et al. Micro-PET imaging of alphavbeta3-integrin expression with 18F-labeled dimeric RGD peptide. Mol Imaging. 2004;3:96–104.
226. Zhang X, Xiong Z, Wu Y, et al. Quantitative PET Imaging of Tumor Integrin {alpha}v{beta}3 Expression with 18F-FRGD2. J Nucl Med. 2006;47:113–121.
227. Chen X, Liu S, Hou Y, et al. MicroPET imaging of breast cancer alphav-integrin expression with 64Cu-labeled dimeric RGD peptides. Mol Imaging Biol. 2004;6: 350–359.
228. Wu Y, Zhang X, Xiong Z, et al. microPET imaging of glioma integrin {alpha}v{beta}3 expression using (64)Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707–1718.
229. Li ZB, Cai W, Cao Q, et al. (64)Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor alpha(v)beta(3) integrin expression. J Nucl Med. 2007;48:1162–1171.
230. Sancey L, Ardisson V, Riou LM, et al. In vivo imaging of tumour angiogenesis in mice with the alpha(v)beta (3) integrin-targeted tracer (99m)Tc-RAFT-RGD. Eur J Nucl Med Mol Imaging. 2007;34(12):2037–2047.
231. Dijkgraaf I, Rijnders AY, Soede A, et al. Synthesis of DOTA-conjugated multivalent cyclic-RGD peptide dendrimers via 1,3-dipolar cycloaddition and their biological evaluation: Implications for tumor targeting and tumor imaging purposes. Org Biomol Chem. 2007;5:935–944.
232. Wadas TJ, Deng H, Sprague JE, et al. Targeting the alphavbeta3 integrin for small-animal PET/CT of osteolytic bone metastases. J Nucl Med. 2009;50:1873–1880.
233. Battle MR, Goggi JL, Allen L, et al. Monitoring tumor response to antiangiogenic sunitinib therapy with 18F-fluciclatide, an 18F-labeled alphaVbeta3-integrin and alphaV beta5-integrin imaging agent. J Nucl Med. 2011;52:424–430.
234. de Bouard S, Herlin P, Christensen JG, et al. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007;9: 412–423.
235. Morrison M, Cuthbertson A. Integrin imaging to evaluate treatment response. Theranostics. 2011;1:149–153.
236. Kenny LM, Coombes RC, Oulie I, et al. Phase I trial of the positron-emitting Arg-Gly-Asp (RGD) peptide radioligand 18F-AH111585 in breast cancer patients. J Nucl Med. 2008;49:879–886.
237. McParland BJ, Miller MP, Spinks TJ, et al. The biodistribution and radiation dosimetry of the Arg-Gly-Asp peptide 18F-AH111585 in healthy volunteers. J Nucl Med. 2008;49:1664–1667.
238. Tomasi G, Kenny L, Mauri F, et al. Quantification of receptor-ligand binding with [(1)F]fluciclatide in metastatic breast cancer patients. Eur J Nucl Med Mol Imaging. 2011;38:2186–2197.
239. Schnell O, Krebs B, Carlsen J, et al. Imaging of integrin {alpha}v{beta}3 expression in patients with malignant glioma by [18F]galacto-RGD positron emission tomography. Neuro Oncol. 2009;11(6):861–870.
240. Doss M, Alpaugh RK, Yu JQ. Biodistribution and radiation dosimetry of angiogenesis marker [18F]RGD-K5 measured using human PET. J Nucl Med. 2009; 50(suppl 2):447.
241. Zhang J. Efficacy study of [18F]RGD-K5 Positron Emission Tomography (PET) as a tool to monitor response to an anti-angiogenic drug (K5-101). ClinicalTrialsgov. 2009;October 1, 2009 ed: U.S. National Institute of Health.
242. Fanti S, Farsad M, Mansi L. PET-CT Beyond FDG: A Quick Guide to Image Interpretation. 1st ed. Berlin: Springer-Verlag; 2010.
243. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803.
244. Roodhart JM, Langenberg MH, Witteveen E, et al. The molecular basis of class side effects due to treatment with inhibitors of the VEGF/VEGFR pathway. Curr Clin Pharmacol. 2008;3:132–143.
245. Ferrara N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr Rev. 2004;25:581–611.
246. Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–1326.
247. Underiner TL, Mallamo JP, Singh J. Syntheses of C12,N13 heterocyclic bridged fused indenopyrrolocarbazoles. J Org Chem. 2002;67:3235–3241.
248. Yuan F, Chen Y, Dellian M, et al. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci U S A. 1996;93:14765–14770.
249. Wang Y, Fei D, Vanderlaan M, et al. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335–345.
250. Hsu AR, Chen X. Advances in anatomic, functional, and molecular imaging of angiogenesis. J Nucl Med. 2008;49:511–514.
251. Nayak TK, Garmestani K, Baidoo KE, et al. PET imaging of tumor angiogenesis in mice with VEGF-A-targeted (86)Y-CHX-A”-DTPA-bevacizumab. Int J Cancer. 2011;128:920–926.
252. Collingridge DR, Carroll VA, Glaser M, et al. The development of [(124)I]iodinated-VG76e: A novel tracer for imaging vascular endothelial growth factor in vivo using positron emission tomography. Cancer Res. 2002;62:5912–5919.
253. Scheer MG, Stollman TH, Boerman OC, et al. Imaging liver metastases of colorectal cancer patients with radiolabelled bevacizumab: Lack of correlation with VEGF-A expression. Eur J Cancer. 2008;44:1835–1840.
254. Eder M, Krivoshein AV, Backer M, et al. ScVEGF-PEG-HBED-CC and scVEGF-PEG-NOTA conjugates: Comparison of easy-to-label recombinant proteins for [68Ga]PET imaging of VEGF receptors in angiogenic vasculature. Nucl Med Biol. 2010;37:405–412.
255. Nagengast WB, de Korte MA, Oude Munnink TH, et al. 89Zr-bevacizumab PET of early antiangiogenic tumor response to treatment with HSP90 inhibitor NVP-AUY922. J Nucl Med. 2010;51:761–767.
256. Nagengast WB, Lub-de Hooge MN, Oosting SF, et al. VEGF-PET imaging is a noninvasive biomarker showing differential changes in the tumor during sunitinib treatment. Cancer Res. 2011;71:143–153.
257. Niu G, Chen X. PET imaging of angiogenesis. PET Clin. 2009;4:17–38.
258. Li S, Peck-Radosavljevic M, Kienast O, et al. Iodine-123-vascular endothelial growth factor-165 (123I-VEGF165). Biodistribution, safety and radiation dosimetry in patients with pancreatic carcinoma. Q J Nucl Med Mol Imaging. 2004; 48:198–206.
259. Fuh G, Garcia KC, de Vos AM. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem. 2000;275:26690–26695.
260. Chen K, Cai W, Li ZB, et al. Quantitative PET imaging of VEGF receptor expression. Mol Imaging Biol. 2009;11:15–22.
261. Blankenberg FG, Backer MV, Levashova Z, et al. In vivo tumor angiogenesis imaging with site-specific labeled (99m)Tc-HYNIC-VEGF. Eur J Nucl Med Mol Imaging. 2006;33:841–848.
262. Backer MV, Levashova Z, Patel V, et al. Molecular imaging of VEGF receptors in angiogenic vasculature with single-chain VEGF-based probes. Nat Med. 2007; 13:504–509.
263. Lu E, Wagner WR, Schellenberger U, et al. Targeted in vivo labeling of receptors for vascular endothelial growth factor: Approach to identification of ischemic tissue. Circulation. 2003;108:97–103.
264. Keyt BA, Nguyen HV, Berleau LT, et al. Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors. Generation of receptor-selective VEGF variants by site-directed mutagenesis. J Biol Chem. 1996;271:5638–5646.
265. Wang H, Cai W, Chen K, et al. A new PET tracer specific for vascular endothelial growth factor receptor 2. Eur J Nucl Med Mol Imaging. 2007;34:2001–2010.
266. Rodriguez-Porcel M, Cai W, Gheysens O, et al. Imaging of VEGF receptor in a rat myocardial infarction model using PET. J Nucl Med. 2008;49:667–673.
267. Blankenberg FG, Mandl S, Cao YA, et al. Tumor imaging using a standardized radiolabeled adapter protein docked to vascular endothelial growth factor. J Nucl Med. 2004;45:1373–1380.
268. Li S, Peck-Radosavljevic M, Kienast O, et al. Imaging gastrointestinal tumours using vascular endothelial growth factor-165 (VEGF165) receptor scintigraphy. Ann Oncol. 2003;14:1274–1277.
269. Li S, Peck-Radosavljevic M, Koller E, et al. Characterization of (123)I-vascular endothelial growth factor-binding sites expressed on human tumour cells: Possible implication for tumour scintigraphy. Int J Cancer. 2001;91:789–796.
270. Cai W, Guzman R, Hsu AR, et al. Positron emission tomography imaging of poststroke angiogenesis. Stroke. 2009;40:270–277.
271. Blankenberg FG, Levashova Z, Sarkar SK, et al. Noninvasive assessment of tumor VEGF receptors in response to treatment with pazopanib: A molecular imaging study. Transl Oncol. 2010;3:56–64.
272. Curran S, Murray GI. Matrix metalloproteinases: Molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer. 2000;36:1621–1630.
273. Hidalgo M, Eckhardt SG. Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst. 2001;93:178–193.
274. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174.
275. Decock J, Thirkettle S, Wagstaff L, et al. Matrix metalloproteinases: Protective roles in cancer. J Cell Mol Med. 2011;15:1254–1265.
276. Gomez DE, Alonso DF, Yoshiji H, et al. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur J Cell Biol. 1997;74:111–122.
277. Foda HD, Zucker S. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov Today. 2001;6:478–482.
278. Iwata H, Kobayashi S, Iwase H, et al. Production of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human breast carcinomas. Jpn J Cancer Res. 1996;87:602–611.
279. Nguyen M, Arkell J, Jackson CJ. Human endothelial gelatinases and angiogenesis. Int J Biochem Cell Biol. 2001;33:960–970.
280. Matter A. Tumor angiogenesis as a therapeutic target. Drug Discov Today. 2001; 6:1005–1024.
281. Hartung D, Schafers M, Fujimoto S, et al. Targeting of matrix metalloproteinase activation for noninvasive detection of vulnerable atherosclerotic lesions. Eur J Nucl Med Mol Imaging. 2007;34(suppl 1):S1–S8.
282. Van de Wiele C, Oltenfreiter R. Imaging probes targeting matrix metalloproteinases. Cancer Biother Radiopharm. 2006;21:409–417.
283. Koivunen E, Arap W, Valtanen H, et al. Tumor targeting with a selective gelatinase inhibitor. Nat Biotechnol. 1999;17:768–774.
284. Kuhnast B, Bodenstein C, Haubner R, et al. Targeting of gelatinase activity with a radiolabeled cyclic HWGF peptide. Nucl Med Biol. 2004;31:337–344.
285. Sprague JE, Li WP, Liang K, et al. In vitro and in vivo investigation of matrix metalloproteinase expression in metastatic tumor models. Nucl Med Biol. 2006;33: 227–237.
286. Ujula T, Huttunen M, Luoto P, et al. Matrix metalloproteinase 9 targeting peptides: Syntheses, 68Ga-labeling, and preliminary evaluation in a rat melanoma xenograft model. Bioconjug Chem. 2010;21:1612–1621.
287. Azhdarinia A, Wilganowski N, Robinson H, et al. Characterization of chemical, radiochemical and optical properties of a dual-labeled MMP-9 targeting peptide. Bioorg Med Chem. 2011;19:3769–3776.
288. Chuang CH, Chuang KH, Wang HE, et al. In vivo positron emission tomography imaging of protease activity by generation of a hydrophobic product from a noninhibitory protease substrate. Clin Cancer Res. 2012;18:238–247.
289. Levy DE, Lapierre F, Liang W, et al. Matrix metalloproteinase inhibitors: A structure-activity study. J Med Chem. 1998;41:199–223.
290. Kiyama R, Tamura Y, Watanabe F, et al. Homology modeling of gelatinase catalytic domains and docking simulations of novel sulfonamide inhibitors. J Med Chem. 1999;42:1723–1738.
291. Pelmenschikov V, Siegbahn PE. Catalytic mechanism of matrix metalloproteinases: Two-layered ONIOM study. Inorg Chem. 2002;41:5659–5666.
292. Aranapakam V, Davis JM, Grosu GT, et al. Synthesis and structure-activity relationship of N-substituted 4-arylsulfonylpiperidine-4-hydroxamic acids as novel, orally active matrix metalloproteinase inhibitors for the treatment of osteoarthritis. J Med Chem. 2003;46:2376–2396.
293. Furumoto S, Iwata R, Ido T. Design and synthesis of fluorine-18 labeled matrix metalloproteinase inhibitors for cancer imaging. J Labelled Comp Radiopharm. 2002;45:975–986.
294. Fei X, Zheng QH, Hutchins GD, et al. Synthesis of MMP inhibitor radiotracers [11C]methyl-CGS 27023A and its analogs, new potential PET breast cancer imaging agents. J Labelled Comp Radiopharm. 2002;45:449–470.
295. Fei X, Zheng Q-H, Liu X, et al. Synthesis of MMP inhibitor radiotracer [11C]CGS 25966, a new potential PET tumor imaging agent. J Labelled Comp Radiopharm. 2003;46:343–351.
296. Zheng QH, Fei X, DeGrado TR, et al. Synthesis, biodistribution and micro-PET imaging of a potential cancer biomarker carbon-11 labeled MMP inhibitor (2R)-2-[[4-(6-fluorohex-1-ynyl)phenyl]sulfonylamino]-3-methylbutyric acid [(11)C]methyl ester. Nucl Med Biol.2003;30:753–760.
297. Zheng QH, Fei X, Liu X, et al. Synthesis and preliminary biological evaluation of MMP inhibitor radiotracers [11C]methyl-halo-CGS 27023A analogs, new potential PET breast cancer imaging agents. Nucl Med Biol. 2002;29:761–770.
298. Kuhnast B, Bodenstein C, Wester HJ, et al. Carbon-11 labelling of a N-sulfonylamino acid derivative: A potential tracer for MMP-2 and MMP-9 imaging. J Labelled Comp Radiopharm. 2003;46:1093–1103.
299. Fei X, Zheng QH, Liu X, et al. Synthesis of radiolabeled biphenylsulfonamide matrix metalloproteinase inhibitors as new potential PET cancer imaging agents. Bioorg Med Chem Lett. 2003;13:2217–2222.
300. Oltenfreiter R, Staelens L, Lejeune A, et al. New radioiodinated carboxylic and hydroxamic matrix metalloproteinase inhibitor tracers as potential tumor imaging agents. Nucl Med Biol. 2004;31:459–468.
301. Oltenfreiter R, Staelens L, Hillaert U, et al. Synthesis, radiosynthesis, in vitro and preliminary in vivo evaluation of biphenyl carboxylic and hydroxamic matrix metalloproteinase (MMP) inhibitors as potential tumor imaging agents. Appl Radiat Isot. 2005;62:903–913.
302. Wagner S, Breyholz HJ, Law MP, et al. Novel fluorinated derivatives of the broad-spectrum MMP inhibitors N-hydroxy-2(R)-[[(4-methoxyphenyl)sulfonyl](benzyl)- and (3-picolyl)-amino]-3-methyl-butanamide as potential tools for the molecular imaging of activated MMPs with PET. J Med Chem.2007;50:5752–5764.
303. Breyholz HJ, Wagner S, Faust A, et al. Radiofluorinated pyrimidine-2,4,6-triones as molecular probes for noninvasive MMP-targeted imaging. ChemMedChem. 2010;5:777–789.
304. Schrigten D, Breyholz HJ, Wagner S, et al. A new generation of radiofluorinated pyrimidine-2,4,6-triones as MMP-targeted radiotracers for positron emission tomography. J Med Chem. 2012;55:223–232.
305. Zheng QH, Fei X, Liu X, et al. Comparative studies of potential cancer biomarkers carbon-11 labeled MMP inhibitors (S)-2-(4′-[11C]methoxybiphenyl-4-sulfonylamino)-3-methylbutyric acid and N-hydroxy-(R)-2-[[(4′-[11C]methoxyphenyl)sulfonyl]benzylamino]-3-methylbut anamide. Nucl Med Biol.2004;31: 77–85.
306. Oltenfreiter R, Staelens L, Labied S, et al. Tryptophane-based biphenylsulfonamide matrix metalloproteinase inhibitors as tumor imaging agents. Cancer Biother Radiopharm. 2005;20:639–647.
307. auf dem Keller U, Bellac CL, Li Y, et al. Novel matrix metalloproteinase inhibitor [18F]marimastat-aryltrifluoroborate as a probe for in vivo positron emission tomography imaging in cancer. Cancer Res. 2010;70:7562–7569.
308. Su H, Spinale FG, Dobrucki LW, et al. Noninvasive targeted imaging of matrix metalloproteinase activation in a murine model of postinfarction remodeling. Circulation. 2005;112:3157–3167.
309. Razavian M, Tavakoli S, Zhang J, et al. Atherosclerosis plaque heterogeneity and response to therapy detected by in vivo molecular imaging of matrix metalloproteinase activation. J Nucl Med. 2011;52:1795–1802.
310. Zhang J, Nie L, Razavian M, et al. Molecular imaging of activated matrix metalloproteinases in vascular remodeling. Circulation. 2008;118:1953–1960.
311. Hynes R. Molecular biology of fibronectin. Annu Rev Cell Biol. 1985;1:67–90.
312. Castellani P, Dorcaratto A, Pau A, et al. The angiogenesis marker ED-B+ fibronectin isoform in intracranial meningiomas. Acta Neurochir (Wien). 2000;142: 277–282.
313. Neri D, Carnemolla B, Nissim A, et al. Targeting by affinity-matured recombinant antibody fragments of an angiogenesis associated fibronectin isoform. Nat Biotechnol. 1997;15:1271–1275.
314. Santimaria M, Moscatelli G, Viale GL, et al. Immunoscintigraphic detection of the ED-B domain of fibronectin, a marker of angiogenesis, in patients with cancer. Clin Cancer Res. 2003;9:571–579.
315. Berndorff D, Borkowski S, Moosmayer D, et al. Imaging of tumor angiogenesis using 99mTc-labeled human recombinant anti-ED-B fibronectin antibody fragments. J Nucl Med. 2006;47:1707–1716.
316. Borsi L, Balza E, Bestagno M, et al. Selective targeting of tumoral vasculature: Comparison of different formats of an antibody (L19) to the ED-B domain of fibronectin. Int J Cancer. 2002;102:75–85.
317. Berndorff D, Borkowski S, Sieger S, et al. Radioimmunotherapy of solid tumors by targeting extra domain B fibronectin: Identification of the best-suited radioimmunoconjugate. Clin Cancer Res. 2005;11:7053s–7063s.
318. Tijink BM, Perk LR, Budde M, et al. (124)I-L19-SIP for immuno-PET imaging of tumour vasculature and guidance of (131)I-L19-SIP radioimmunotherapy. Eur J Nucl Med Mol Imaging. 2009;36:1235–1244.
319. Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999; 20:157–198.
320. Oberg KE, Reubi JC, Kwekkeboom DJ, et al. Role of somatostatins in gastroenteropancreatic neuroendocrine tumor development and therapy. Gastroenterology. 2010; 139:742–753, 53 e1.
321. Hicks RJ. Use of molecular targeted agents for the diagnosis, staging and therapy of neuroendocrine malignancy. Cancer Imaging. 2010;10(Spec no A): S83–S91.
322. Gabriel M, Decristoforo C, Donnemiller E, et al. An intrapatient comparison of 99mTc-EDDA/HYNIC-TOC with 111In-DTPA-octreotide for diagnosis of somatostatin receptor-expressing tumors. J Nucl Med. 2003;44:708–716.
323. Decristoforo C, Pickett RD, Verbruggen A. Feasibility and availability of (68)Ga-labelled peptides. Eur J Nucl Med Mol Imaging. 2012;39(suppl 1):S31–S40.
324. Henze M, Schuhmacher J, Hipp P, et al. PET imaging of somatostatin receptors using [68GA]DOTA-D-Phe1-Tyr3-octreotide: First results in patients with meningiomas. J Nucl Med. 2001;42:1053–1056.
325. Loktionova NS, Belozub AN, Filosofov DV, et al. Improved column-based radiochemical processing of the generator produced 68Ga. Appl Radiat Isot. 2011;69: 942–946.
326. Breeman WA, de Blois E, Sze Chan H, et al. (68)Ga-labeled DOTA-peptides and (68)Ga-labeled radiopharmaceuticals for positron emission tomography: Current status of research, clinical applications, and future perspectives. Semin Nucl Med. 2011;41:314–321.
327. Traub-Weidinger T, Von Guggenberg E, Dobrozemsky G, et al. Preliminary experience with (68)Ga-DOTA-lanreotide positron emission tomography. Q J Nucl Med Mol Imaging. 2010;54:52–60.
328. Hanaoka H, Tominaga H, Yamada K, et al. Evaluation of (64)Cu-labeled DOTA-D-Phe(1)-Tyr (3)-octreotide ((64)Cu-DOTA-TOC) for imaging somatostatin receptor-expressing tumors. Ann Nucl Med. 2009;23:559–567.
329. Anderson CJ, Dehdashti F, Cutler PD, et al. 64Cu-TETA-octreotide as a PET imaging agent for patients with neuroendocrine tumors. J Nucl Med. 2001;42: 213–221.
330. Meisetschlager G, Poethko T, Stahl A, et al. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: Biodistribution and diagnostic evaluation compared with [111In]DTPA-octreotide. J Nucl Med. 2006;47:566–573.
331. Haubner R. PET radiopharmaceuticals in radiation treatment planning - synthesis and biological characteristics. Radiother Oncol. 2010;96:280–287.
332. Ginj M, Zhang H, Waser B, et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci U S A. 2006;103:16436–16441.
333. Giovacchini G, Nicolas G, Forrer F. Peptide receptor radionuclide therapy with somatostatin analogues in neuroendocrine tumors. Anticancer Agents Med Chem. 2012.
334. Petrik M, Knetsch PA, Knopp R, et al. Radiolabelling of peptides for PET, SPECT and therapeutic applications using a fully automated disposable cassette system. Nucl Med Commun. 2011;32:887–895.
335. Li X, Huang J, Yi P, et al. Single-chain estrogen receptors (ERs) reveal that the ERalpha/beta heterodimer emulates functions of the ERalpha dimer in genomic estrogen signaling pathways. Mol Cell Biol. 2004;24:7681–7694.
336. Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006; 116:561–570.
337. Sundararajan L, Linden HM, Link JM, et al. 18F-Fluoroestradiol. Semin Nucl Med. 2007;37:470–476.
338. Yoo J, Dence CS, Sharp TL, et al. Synthesis of an estrogen receptor beta-selective radioligand: 5-[18F]fluoro-(2R,3S)-2,3-bis(4-hydroxyphenyl)pentanenitrile and comparison of in vivo distribution with 16alpha-[18F]fluoro-17beta-estradiol. J Med Chem. 2005;48:6366–6378.
339. Jonson SD, Bonasera TA, Dehdashti F, et al. Comparative breast tumor imaging and comparative in vitro metabolism of 16alpha-[18F]fluoroestradiol-17beta and 16beta-[18F]fluoromoxestrol in isolated hepatocytes. Nucl Med Biol. 1999;26: 123–130.
340. Zlotnik A. New insights on the role of CXCR4 in cancer metastasis. J Pathol. 2008;215:211–213.
341. Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11:597–606.
342. Woodard LE, Nimmagadda S. CXCR4-based imaging agents. J Nucl Med. 2011;52:1665–1669.
343. De Silva RA, Peyre K, Pullambhatla M, et al. Imaging CXCR4 expression in human cancer xenografts: Evaluation of monocyclam 64Cu-AMD3465. J Nucl Med. 2011;52:986–993.
344. Tamamura H, Xu Y, Hattori T, et al. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: A strong anti-HIV peptide T140. Biochem Biophys Res Commun. 1998;253:877–882.
345. Tamamura H, Tsutsumi H, Masuno H, et al. Development of low molecular weight CXCR4 antagonists by exploratory structural tuning of cyclic tetra- and pentapeptide-scaffolds towards the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Curr Med Chem. 2007;14:93–102.
346. Dessinioti C, Antoniou C, Katsambas A, et al. Melanocortin 1 receptor variants: Functional role and pigmentary associations. Photochem Photobiol. 2011;87: 978–987.
347. Sawyer TK, Sanfilippo PJ, Hruby VJ, et al. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: A highly potent alpha-melanotropin with ultralong biological activity. Proc Natl Acad Sci U S A. 1980;77:5754–5758.
348. Giblin MF, Wang N, Hoffman TJ, et al. Design and characterization of alpha-melanotropin peptide analogs cyclized through rhenium and technetium metal coordination. Proc Natl Acad Sci U S A. 1998;95:12814–12818.
349. Cody WL, Mahoney M, Knittel JJ, et al. Cyclic melanotropins. 9. 7-D-Phenylalanine analogues of the active-site sequence. J Med Chem. 1985;28:583–588.
350. Al-Obeidi F, Castrucci AM, Hadley ME, et al. Potent and prolonged acting cyclic lactam analogues of alpha-melanotropin: Design based on molecular dynamics. J Med Chem. 1989;32:2555–2561.
351. Cheng Z, Xiong Z, Subbarayan M, et al. 64Cu-labeled alpha-melanocyte-stimulating hormone analog for microPET imaging of melanocortin 1 receptor expression. Bioconjug Chem. 2007;18:765–772.
352. Wei L, Miao Y, Gallazzi F, et al. Gallium-68-labeled DOTA-rhenium-cyclized alpha-melanocyte-stimulating hormone analog for imaging of malignant melanoma. Nucl Med Biol. 2007;34:945–953.
353. Cantorias MV, Figueroa SD, Quinn TP, et al. Development of high-specific-activity (68)Ga-labeled DOTA-rhenium-cyclized alpha-MSH peptide analog to target MC1 receptors overexpressed by melanoma tumors. Nucl Med Biol. 2009;36: 505–513.
354. Froidevaux S, Calame-Christe M, Schuhmacher J, et al. A gallium-labeled DOTA-alpha-melanocyte-stimulating hormone analog for PET imaging of melanoma metastases. J Nucl Med. 2004;45:116–123.
355. Miao Y, Owen NK, Whitener D, et al. In vivo evaluation of 188Re-labeled alpha-melanocyte stimulating hormone peptide analogs for melanoma therapy. Int J Cancer. 2002;101:480–487.
356. Miao Y, Shelton T, Quinn TP. Therapeutic efficacy of a 177Lu-labeled DOTA conjugated alpha-melanocyte-stimulating hormone peptide in a murine melanoma-bearing mouse model. Cancer Biother Radiopharm. 2007;22:333–341.
357. Quartara L, Maggi CA. The tachykinin NK1 receptor. Part II: Distribution and pathophysiological roles. Neuropeptides. 1998;32:1–49.
358. Hennig IM, Laissue JA, Horisberger U, et al. Substance-P receptors in human primary neoplasms: Tumoral and vascular localization. Int J Cancer. 1995;61: 786–792.
359. Vanderhaeghen JJ, Signeau JC, Gepts W. New peptide in the vertebrate CNS reacting with antigastrin antibodies. Nature. 1975;257:604–605.
360. Roosenburg S, Laverman P, van Delft FL, et al. Radiolabeled CCK/gastrin peptides for imaging and therapy of CCK2 receptor-expressing tumors. Amino Acids. 2011;41:1049–1058.
361. Reubi JC, Waser B. Unexpected high incidence of cholecystokinin-B/gastrin receptors in human medullary thyroid carcinomas. Int J Cancer. 1996;67:644–647.
362. Behr TM, Jenner N, Behe M, et al. Radiolabeled peptides for targeting cholecystokinin-B/gastrin receptor-expressing tumors. J Nucl Med. 1999;40:1029–1044.
363. Sosabowski JK, Matzow T, Foster JM, et al. Targeting of CCK-2 receptor-expressing tumors using a radiolabeled divalent gastrin peptide. J Nucl Med. 2009;50:2082–2089.
364. Aloj L, Caraco C, Panico M, et al. In vitro and in vivo evaluation of 111In-DTPAGlu-G-CCK8 for cholecystokinin-B receptor imaging. J Nucl Med. 2004;45: 485–494.
365. Chakder S, Rattan S. The entire vasoactive intestinal polypeptide molecule is required for the activation of the vasoactive intestinal polypeptide receptor: Functional and binding studies on opossum internal anal sphincter smooth muscle. J Pharmacol Exp Ther. 1993;266:392–399.
366. Raderer M, Kurtaran A, Leimer M, et al. Value of peptide receptor scintigraphy using (123)I-vasoactive intestinal peptide and (111)In-DTPA-D-Phe1-octreotide in 194 carcinoid patients: Vienna University Experience, 1993 to 1998. J Clin Oncol. 2000;18:1331–1336.
367. Cheng D, Yin D, Li G, et al. Radiolabeling and in vitro and in vivo characterization of [18F]FB-[R(8,15,21), L17]-VIP as a PET imaging agent for tumor overexpressed VIP receptors. Chem Biol Drug Des. 2006;68:319–325.
368. Pallela VR, Thakur ML, Chakder S, et al. 99mTc-labeled vasoactive intestinal peptide receptor agonist: Functional studies. J Nucl Med. 1999;40:352–360.
369. Thakur ML, Aruva MR, Gariepy J, et al. PET imaging of oncogene overexpression using 64Cu-vasoactive intestinal peptide (VIP) analog: Comparison with 99mTc-VIP analog. J Nucl Med. 2004;45:1381–1389.
370. Rao PS, Thakur ML, Pallela V, et al. 99mTc labeled VIP analog: Evaluation for imaging colorectal cancer. Nucl Med Biol. 2001;28:445–450.
371. Thakur ML, Marcus CS, Saeed S, et al. 99mTc-labeled vasoactive intestinal peptide analog for rapid localization of tumors in humans. J Nucl Med. 2000;41: 107–110.
372. Jagoda EM, Aloj L, Seidel J, et al. Comparison of an 18F labeled derivative of vasoactive intestinal peptide and 2-deoxy-2-[18F]fluoro-D-glucose in nude mice bearing breast cancer xenografts. Mol Imaging Biol. 2002;4:369–379.
373. Sunday ME, Kaplan LM, Motoyama E, et al. Gastrin-releasing peptide (mammalian bombesin) gene expression in health and disease. Lab Invest. 1988;59: 5–24.
374. Spindel ER, Giladi E, Segerson TP, et al. Bombesin-like peptides: Of ligands and receptors. Recent Prog Horm Res. 1993;48:365–391.
375. Smith CJ, Volkert WA, Hoffman TJ. Gastrin releasing peptide (GRP) receptor targeted radiopharmaceuticals: A concise update. Nucl Med Biol. 2003;30:861–868.
376. Markwalder R, Reubi JC. Gastrin-releasing peptide receptors in the human prostate: Relation to neoplastic transformation. Cancer Res. 1999;59:1152–1159.
377. Sun B, Schally AV, Halmos G. The presence of receptors for bombesin/GRP and mRNA for three receptor subtypes in human ovarian epithelial cancers. Regul Pept. 2000;90:77–84.
378. Rogers BE, Rosenfeld ME, Khazaeli MB, et al. Localization of iodine-125-mIP-Des-Met14-bombesin (7-13)NH2 in ovarian carcinoma induced to express the gastrin releasing peptide receptor by adenoviral vector-mediated gene transfer. J Nucl Med. 1997;38:1221–1229.
379. Moody TW, Pert CB, Rivier J, et al. Bomebesin: Specific binding to rat brain membranes. Proc Natl Acad Sci U S A. 1978;75:5372–5376.
380. Coy DH, Jensen RT, Jiang NY, et al. Systematic development of bombesin/gastrin-releasing peptide antagonists. J Natl Cancer Inst Monogr. 1992:133–139.
381. Zalutsky MR, Narula AS. A method for the radiohalogenation of proteins resulting in decreased thyroid uptake of radioiodine. Int J Rad Appl Instrum A. 1987;38: 1051–1055.
382. Karra SR, Schibli R, Gali H, et al. 99mTc-labeling and in vivo studies of a bombesin analogue with a novel water-soluble dithiadiphosphine-based bifunctional chelating agent. Bioconjug Chem. 1999;10:254–260.
383. Van de Wiele C, Dumont F, Vanden Broecke R, et al. Technetium-99m RP527, a GRP analogue for visualisation of GRP receptor-expressing malignancies: A feasibility study. Eur J Nucl Med. 2000;27:1694–1699.
384. Van de Wiele C, Dumont F, Dierckx RA, et al. Biodistribution and dosimetry of (99m)Tc-RP527, a gastrin-releasing peptide (GRP) agonist for the visualization of GRP receptor-expressing malignancies. J Nucl Med. 2001;42:1722–1727.
385. Baidoo KE, Lin KS, Zhan Y, et al. Design, synthesis, and initial evaluation of high-affinity technetium bombesin analogues. Bioconjug Chem. 1998;9:218–225.
386. Nock B, Nikolopoulou A, Chiotellis E, et al. [99mTc]Demobesin 1, a novel potent bombesin analogue for GRP receptor-targeted tumour imaging. Eur J Nucl Med Mol Imaging. 2003;30:247–258.
387. La Bella R, Garcia-Garayoa E, Bahler M, et al. A 99mTc(I)-postlabeled high affinity bombesin analogue as a potential tumor imaging agent. Bioconjug Chem. 2002;13:599–604.
388. La Bella R, Garcia-Garayoa E, Langer M, et al. In vitro and in vivo evaluation of a 99mTc(I)-labeled bombesin analogue for imaging of gastrin releasing peptide receptor-positive tumors. Nucl Med Biol. 2002;29:553–560.
389. Breeman WA, de Jong M, Erion JL, et al. Preclinical comparison of (111)In-labeled DTPA- or DOTA-bombesin analogs for receptor-targeted scintigraphy and radionuclide therapy. J Nucl Med. 2002;43:1650–1656.
390. Rogers BE, Manna DD, Safavy A. In vitro and in vivo evaluation of a 64Cu-labeled polyethylene glycol-bombesin conjugate. Cancer Biother Radiopharm. 2004;19:25–34.
391. Scheffel U, Pomper MG. PET imaging of GRP receptor expression in prostate cancer. J Nucl Med. 2004;45:1277–1278.
392. Bass LA, Wang M, Welch MJ, et al. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug Chem. 2000;11: 527–532.
393. Rogers BE, Anderson CJ, Connett JM, et al. Comparison of four bifunctional chelates for radiolabeling monoclonal antibodies with copper radioisotopes: Biodistribution and metabolism. Bioconjug Chem. 1996;7:511–522.
394. Chen X, Park R, Hou Y, et al. microPET and autoradiographic imaging of GRP receptor expression with 64Cu-DOTA-[Lys3]bombesin in human prostate adenocarcinoma xenografts. J Nucl Med. 2004;45:1390–1397.
395. Zhang H, Schuhmacher J, Waser B, et al. DOTA-PESIN, a DOTA-conjugated bombesin derivative designed for the imaging and targeted radionuclide treatment of bombesin receptor-positive tumours. Eur J Nucl Med Mol Imaging. 2007; 34:1198–1208.
396. Abiraj K, Mansi R, Tamma ML, et al. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J Nucl Med. 2011;52:1970–1978.
397. Schroeder RP, van Weerden WM, Krenning EP, et al. Gastrin-releasing peptide receptor-based targeting using bombesin analogues is superior to metabolism-based targeting using choline for in vivo imaging of human prostate cancer xenografts. Eur J Nucl Med Mol Imaging.2011;38:1257–1266.
398. Mansi R, Wang X, Forrer F, et al. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur J Nucl Med Mol Imaging. 2011;38:97–107.
399. Lears KA, Ferdani R, Liang K, et al. In vitro and in vivo evaluation of 64Cu-labeled SarAr-bombesin analogs in gastrin-releasing peptide receptor-expressing prostate cancer. J Nucl Med. 2011;52:470–477.
400. Maecke HR, Hofmann M, Haberkorn U. (68)Ga-labeled peptides in tumor imaging. J Nucl Med. 2005;46(suppl 1):172S–178S.
401. Zhang X, Cai W, Cao F, et al. 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J Nucl Med. 2006;47:492–501.
402. Dimitrakopoulou-Strauss A, Hohenberger P, Haberkorn U, et al. 68Ga-labeled bombesin studies in patients with gastrointestinal stromal tumors: Comparison with 18F-FDG. J Nucl Med. 2007;48:1245–1250.
403. Dimitrakopoulou-Strauss A, Seiz M, Tuettenberg J, et al. Pharmacokinetic studies of Ga-labeled Bombesin (Ga-BZH) and F-18 FDG PET in patients with recurrent gliomas and comparison to grading: Preliminary results. Clin Nucl Med. 2011;36:101–108.
404. Hohne A, Mu L, Honer M, et al. Synthesis, 18F-labeling, and in vitro and in vivo studies of bombesin peptides modified with silicon-based building blocks. Bioconjug Chem. 2008;19:1871–1879.
405. Yang M, Gao H, Zhou Y, et al. F-labeled GRPR agonists and antagonists: A comparative study in prostate cancer imaging. Theranostics. 2011;1:220–229.
406. Mu L, Honer M, Becaud J, et al. In vitro and in vivo characterization of novel 18F-labeled bombesin analogues for targeting GRPR-positive tumors. Bioconjug Chem. 2010;21:1864–1871.
407. Jackson AB, Nanda PK, Rold TL, et al. (64)Cu-NO2A-RGD-Glu-6-Ahx-BBN(7-14)NH(2): A heterodimeric targeting vector for positron emission tomography imaging of prostate cancer. Nucl Med Biol. 2012:39(3):377–387.
408. Liu Z, Yan Y, Liu S, et al. (18)F, (64)Cu, and (68)Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug Chem. 2009;20:1016–1025.
409. Liu Z, Li ZB, Cao Q, et al. Small-animal PET of tumors with (64)Cu-labeled RGD-bombesin heterodimer. J Nucl Med. 2009;50:1168–1177.
410. Carraway R, Leeman SE. The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J Biol Chem. 1973;248:6854–6861.
411. Nemeroff CB, Bissette G, Manberg PJ, et al. Neurotensin-induced hypothermia: Evidence for an interaction with dopaminergic systems and the hypothalamic–pituitary–thyroid axis. Brain Res. 1980;195:69–84.
412. Vincent JP. Neurotensin receptors: Binding properties, transduction pathways, and structure. Cell Mol Neurobiol. 1995;15:501–512.
413. Kitabgi P. Effects of neurotensin on intestinal smooth muscle: Application to the study of structure–activity relationships. Ann N Y Acad Sci. 1982;400:37–55.
414. Tyler BM, Cusack B, Douglas CL, et al. Evidence for additional neurotensin receptor subtypes: Neurotensin analogs that distinguish between neurotensin-mediated hypothermia and antinociception. Brain Res. 1998;792:246–252.
415. Labbe-Jullie C, Dubuc I, Brouard A, et al. In vivo and in vitro structure-activity studies with peptide and pseudopeptide neurotensin analogs suggest the existence of distinct central neurotensin receptor subtypes. J Pharmacol Exp Ther. 1994;268:328–336.
416. Mazella J, Zsurger N, Navarro V, et al. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. J Biol Chem. 1998;273:26273–26276.
417. Reubi JC, Zimmermann A, Jonas S, et al. Regulatory peptide receptors in human hepatocellular carcinomas. Gut. 1999;45:766–774.
418. Reubi JC, Waser B, Friess H, et al. Neurotensin receptors: A new marker for human ductal pancreatic adenocarcinoma. Gut. 1998;42:546–550.
419. Przedborski S, Levivier M, Cadet JL. Neurotensin receptors in human meningiomas. Ann Neurol. 1991;30:650–654.
420. Granier C, van Rietschoten J, Kitabgi P, et al. Synthesis and characterization of neurotensin analogues for structure/activity relationship studies. Acetyl-neurotensin-(8–13) is the shortest analogue with full binding and pharmacological activities. Eur J Biochem. 1982;124:117–124.
421. Myers RM, Shearman JW, Kitching MO, et al. Cancer, chemistry, and the cell: Molecules that interact with the neurotensin receptors. ACS Chem Biol. 2009;4: 503–525.
422. Garcia-Garayoa E, Allemann-Tannahill L, Blauenstein P, et al. In vitro and in vivo evaluation of new radiolabeled neurotensin(8-13) analogues with high affinity for NT1 receptors. Nucl Med Biol. 2001;28:75–84.
423. Chavatte K, Terriere D, Jeannin L, et al. Labelling and evaluation of new stabilised neurotensin (8-13) analogues for single photon emission tomography (SPET). J Labelled Comp Radiopharm. 1999;42:423–435.
424. Garcia-Garayoa E, Blauenstein P, Bruehlmeier M, et al. Preclinical evaluation of a new, stabilized neurotensin(8–13) pseudopeptide radiolabeled with (99m)tc. J Nucl Med. 2002;43:374–383.
425. Garcia-Garayoa E, Maes V, Blauenstein P, et al. Double-stabilized neurotensin analogues as potential radiopharmaceuticals for NTR-positive tumors. Nucl Med Biol. 2006;33:495–503.
426. Bruehlmeier M, Garayoa EG, Blanc A, et al. Stabilization of neurotensin analogues: Effect on peptide catabolism, biodistribution and tumor binding. Nucl Med Biol. 2002;29:321–327.
427. Buchegger F, Bonvin F, Kosinski M, et al. Radiolabeled neurotensin analog, 99mTc-NT-XI, evaluated in ductal pancreatic adenocarcinoma patients. J Nucl Med. 2003;44:1649–1654.
428. Maes V, Garcia-Garayoa E, Blauenstein P, et al. Novel 99mTc-labeled neurotensin analogues with optimized biodistribution properties. J Med Chem. 2006; 49:1833–1836.
429. Maina T, Nikolopoulou A, Stathopoulou E, et al. [99mTc]Demotensin 5 and 6 in the NTS1-R-targeted imaging of tumours: Synthesis and preclinical results. Eur J Nucl Med Mol Imaging. 2007;34:1804–1814.
430. Gabriel M, Decristoforo C, Woll E, et al. [99mTc]demotensin VI: Biodistribution and initial clinical results in tumor patients of a pilot/phase I study. Cancer Biother Radiopharm. 2011;26:557–563.
431. Bergmann R, Scheunemann M, Heichert C, et al. Biodistribution and catabolism of (18)F-labeled neurotensin(8-13) analogs. Nucl Med Biol. 2002;29:61–72.
432. Alshoukr F, Prignon A, Brans L, et al. Novel DOTA-neurotensin analogues for 111In scintigraphy and 68Ga PET imaging of neurotensin receptor-positive tumors. Bioconjug Chem. 2011;22:1374–1385.
433. Kaim AH, Weber B, Kurrer MO, et al. F-18-FDG and F-18-FET uptake in experimental soft tissue infection. Eur J Nucl Med Mol Imaging. 2002;29: 648–654.