Wesley Palatnick
HIGH-YIELD FACTS
• Suspect methemoglobinemia in patients with cyanosis unresponsive to 100% oxygen.
• The antidote for methemoglobinemia is methylene blue.
• In patients with methemoglobinemia, conventional pulse oximetry produces factitiously low oxygen saturations.
• Co-oximetry is the method of choice for methemoglobin measurement.
• Young infants with diarrhea and sepsis can develop methemoglobinemia.
INTRODUCTION
Methemoglobinemia occurs when the ferrous (Fe+2) iron of hemoglobin is oxidized to its ferric (Fe+3) counterpart. The amount in the blood is expressed as a percentage of the total hemoglobin. Because of its decreased affinity for oxygen, hypoxia and a shift of the oxygen dissociation curve to the left results.1,2 Methemoglobinemia may be hereditary or acquired; the former is usually milder, occurs early in life, and may be relatively asymptomatic. The acquired form is often more severe and secondary to a diverse group of chemicals and drugs. The classic presentation of methemoglobinemia is cyanosis that does not correct with the administration of 100% oxygen.1,2
PATHOPHYSIOLOGY
The iron of hemoglobin is ferrous (Fe+2) which is capable of carrying oxygen. However, background oxidative stress converts it to its ferric (Fe+3) counterpart resulting in the formation of small amounts of methemoglobin. It is incapable of carrying oxygen, but the body has protective mechanisms to reverse the process.1–3
The major process is the cytochrome b5 methemoglobin reductase system. This two-enzyme process accounts for 99% of the methemoglobin reduction. This is the NADH methemoglobin reductase in older literature.1–3 Another enzyme that can reduce methemoglobin is reduced nicotinamide adenine dinucleotidephosphate (NADPH)-methemoglobin reductase. This system has negligible activity in normal conditions. However, it has an affinity for dyes such as methylene blue and likely plays a role in metabolizing oxidant xenobiotics.2 Ascorbic acid and glutathione may also play a role in reducing small amounts of methemoglobin.2
Normally, only 1% of hemoglobin is methemoglobin at any time.2,4,5 However, if significant oxidative stress is present, these protective systems may be overcome resulting in significant methemoglobinemia.
ETIOLOGY OF METHEMOGLOBINEMIA
Methemoglobinemia may be either hereditary or acquired. The former is uncommon, presents very early, often in the first hours or days of life, and may be misdiagnosed as congenital cyanotic heart disease. Acquired methemoglobin is more common and is more likely to be severe and life threatening.
Hereditary methemoglobinemia is a consequence of cytochrome b5 reductase deficiency, or the presence of one of a number of abnormal hemoglobin variants termed Hemoglobin M. Deficiency of the reduced nicotinamide adenine dinucleotidephosphate (NADPH)-methemoglobin reductase also occurs, but these patients do not manifest methemoglobinemia as this pathway normally plays a very minor role in methemoglobin reduction.1,2
Patients with cytochrome b5 reductase deficiency may be either homozygous or heterozygous. The former have little or no enzyme activity and rely on other endogenous pathways to reduce methemoglobin. They often have significant percentages of methemoglobin, in the 10–50% range. Despite these high values, these patients are often asymptomatic unless exposed to an oxidant stress. Heterozygous individuals usually have insignificant methemoglobin percentages.1,2
Hemoglobin M refers to a number of abnormal hemoglobin variants that are resistant to the normal erythrocyte mechanisms that reduce the ferric (Fe+3) to the ferrous (Fe+2) state. These disorders are autosomal dominant and present early in life. These patients are cyanotic with methemoglobin percentages in the 25–30% range. There is no effective therapy. Only individuals who have the heterozygous form of the hemoglobin variant are known, as having the homozygous variant is not compatible with life.1,6,7
Acquired methemoglobinemia occurs when an individual is exposed to a heterogeneous group of drugs and chemicals that have the ability to overcome endogenous anti-oxidant mechanisms resulting in the oxidation of hemoglobin to methemoglobin. This may occur by direct oxidation of the hemoglobin molecule or more commonly, indirectly by the production of free radicals which then oxidize hemoglobin to methemoglobin.3,7
Agents that can produce methemoglobinemia are numerous (Table 128-1). However, because of variability in individual metabolism, exposures do not always result in the development of methemoglobinemia.2 The most common methemoglobinemia inducers are dapsone, benzocaine, other local anesthetics, and various forms of nitrites.1,5,6,8 Because of the significant risk of methemoglobinemia, benzocaine and prilocaine should be avoided in the young child.9 Benzocaine-containing teething gels are particularly prone to produce methemoglobinemia because the transmucosal absorption from the oral cavity bypasses first-pass hepatic metabolism.10 Dapsone has been implicated in methemoglobinemia in both overdose and during therapeutic dosing.5,11,12
|
TABLE 128-1 |
Acquired Causes of Methemoglobinemia |

A relatively common, but less understood form of acquired methemoglobinemia occurs in very young infants with diarrhea. While acidosis or sepsis is usually present, significant methemoglobinemia may occur without either. The mechanism is not understood. Contributing factors likely include lower levels of cytochrome b5 reductase and that fetal hemoglobin is more easily oxidized to methemoglobin than adult hemoglobin.2,13
CLINICAL PRESENTATION
The classic clinical presentation is cyanosis that is unresponsive to the administration of supplemental oxygen. In infants, the differential diagnosis includes congenital cyanotic heart disease. This is usually differentiated clinically. Children with cyanosis secondary to cyanotic heart disease have a low oxygen saturation and partial pressure of oxygen despite the administration of supplemental oxygen, while those with methemoglobinemia have normal oxygen saturation and high partial pressures of oxygen on blood gas.7
The ubiquity of pulse oximeters has resulted in another clinical presentation in children without cyanosis. The presence of methemoglobin confounds oxygen saturation measurement by conventional pulse oximeters. This results in factitiously low oxygen saturation readings in an otherwise well-appearing noncyanotic child. An additional clue for the presence of methemoglobin is no improvement of the oxygen saturation after the administration of supplemental oxygen. As methemoglobin increases, cyanosis manifests and the oxygen saturation plateaus at 85%.2,14 Recently, pulse oximeters capable of measuring methemoglobin (and carboxyhemoglobin) have been developed.15,16
The cyanosis of methemoglobinemia has also been described as being browner than blue—so called “chocolate cyanosis.”1 This is most evident in the lips and mucous membranes but may also be seen in the nail beds. However, this may be difficult to distinguish in patients with darker skin tones.
Other symptoms are those related to the functional anemia and relative tissue hypoxia and the severity of symptoms increases with increasing -percentage of methemoglobinemia. A methemoglobin percentage of 20–30% may result in anxiety, lightheadedness, headache, and tachycardia. A value of 30–50% is usually associated with confusion, dizziness, tachypnea, and worsening tachycardia. Methemoglobin percentages greater than 50% typically result in severe morbidity such as coma, seizures, cardiac dysrhythmias, and potentially death.2,4,5 Underlying cardio respiratory illness or anemia can result in more severe symptoms at lower methemoglobin percentages.10
DIAGNOSIS
Blood containing significant methemoglobin concentrations may appear chocolate brown and this can be demonstrated if placed on filter paper at the bedside. The chocolate brown color remains despite exposure to atmospheric oxygen. This is in contrast to the red-violet color of deoxygenated blood that will become more bright red after exposure to oxygen2 (Fig. 128-1).
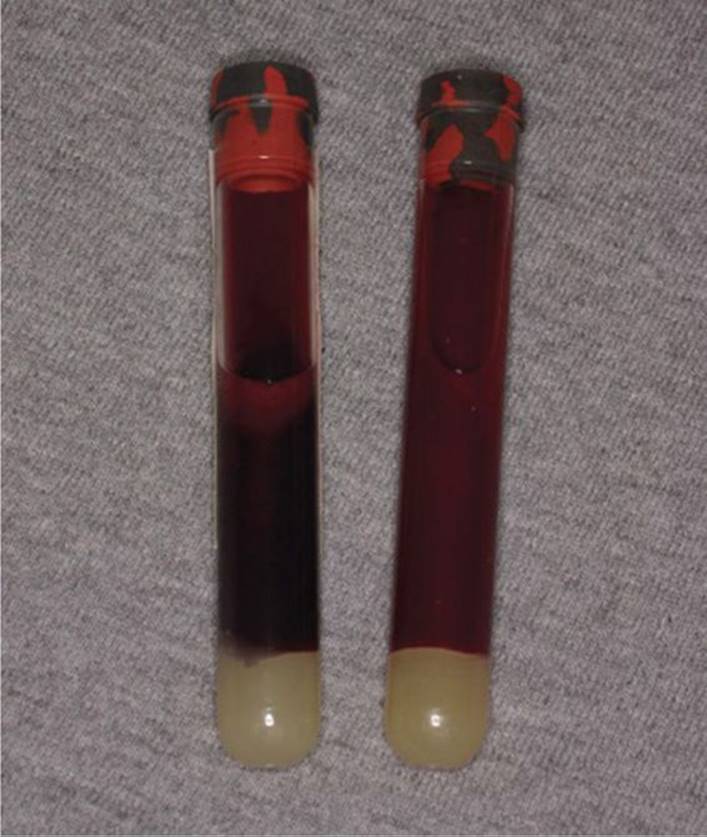
FIGURE 128-1. “Chocolate blood” from an arterial sample of a patient with methemoglobinemia (left) compared to the normal bright red arterial blood (right). (Reproduced with permission from Knoop K. The Atlas of Emergency Medicine, 3rd ed. McGraw-Hill Professional; 2009 (Photo contributor Kevin J. Knoop, MD., MS)).
Pulse oximetry can be helpful in the diagnosis of methemoglobinemia. Conventional units measure light absorbance at two wavelengths (600 and 940 nm) which corresponds to oxygenated and deoxygenated hemoglobin. The presence of methemoglobin interferes and renders the readings factitiously low.2,14 Whether or not cyanosis is present, lack of improvement of oxygen saturation after treatment with supplemental oxygen suggests methemoglobinemia particularly in patients without history of cardiopulmonary disease. As well, the presence of a significant difference between the oxygen saturation measured by pulse oximetry and that calculated on an arterial blood gas (>5%), called a “saturation gap,” is another clue for the presence of methemoglobinemia. Other causes of a “saturation gap” include sulfhemoglobinemia and carboxyhemoglobinemia.2
Measurement of methemoglobin percentage by co-oximetry is the definitive test. It identifies methemoglobin as well as other forms of hemoglobin by spectrophotometry. Because methylene blue has similar absorbance characteristics as methemoglobin, it is important to perform co-oximetry prior to treatment as presence of methylene blue can result in factitiously high methemoglobin readings.14
Recently introduced “pulse co-oximeters” (Masimo Corporation Rainbow Pulse Co-Oximeters) measure up to 12 light wavelengths and can estimate percentages of methemoglobin as well as carboxyhemoglobin and total hemoglobin. These instruments enable measurement of methemoglobin at the bedside, facilitate patient management, and may replace conventional co-oximetry as the definitive test. However, they are not yet in widespread use.15,16
MANAGEMENT
Management of methemoglobinemia consists of general supportive care and antidote administration. As in all emergent situations, the airway, breathing, and circulation must be assessed and managed by routine protocols as required.
Methylene blue is the antidote. It is converted by glucose-6-phosphate dehydrogenase (G6PD) to leukomethylene blue which then reduces methemoglobin (Fe+3) to hemoglobin (Fe+2). The indications for administration of methylene blue are based upon methemoglobin percentages and clinical findings. Treatment is generally recommended for a methemoglobin concentration of 20% or greater in a symptomatic patient and 30% or higher in an asymptomatic patient.1,2 However, treatment should be considered in a symptomatic patient at even lower concentrations.17 The dose of methylene blue is 1 mg/kg of a 1% solution given intravenously. This can be repeated in 30 minutes if required. The maximum dose is 7 mg/kg.1 Methylene blue requires G6PD to exert its effect and may be ineffective in patients with a G6PD deficiency which is more common in patients of African, Asian, Middle Eastern, or Mediterranean descent.4
Potential adverse effects of methylene blue include a paradoxical increase in methemoglobinemia and hemolytic anemia. The latter may occur with the administration of high doses (>7 mg/kg)1 in patients with dapsone-induced methemoglobinemia and in G6PD-deficient patients.4
Other therapies that have been tried include exchange transfusion, hyperbaric oxygen, and the administration of ascorbic acid.6
DISPOSITION
Patients with methemoglobinemia should be admitted to hospital to monitor for recurrences.
REFERENCES
1. Curry S. Methemoglobinemia. Ann Emerg Med. 1982;11:214.
2. Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med. 1999; 34:646.
3. Umbreit J. Methemoglobin- it’s not just blue: a concise review. Am J Hematol. 2007;82:134.
4. Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J. 2011;104:757.
5. Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia. A retrospective series of 138 cases at 2 teaching hospitals. Medicine. 2004;83:265.
6. Hall, AH, Kulig KW, Rumack BH. Drug- and chemical-induced methemoglobinemia. Clinical features and management. Med Toxicol. 1986;1:253.
7. Rehmen HU. Methemoglobinemia. West J Med. 2001;175:193.
8. Bradberry SM. Occupational methemoglobinemia. Mechanisms of production, features, diagnosis and management including the use of methylene blue. Toxicol Rev. 2003;22:13.
9. Guay J. Methemoglobinemia related to local anesthetics: a summary of 242 episodes. Anesth Analg. 2009;108:837.
10. Gentile DA. Severe methemoglobinemia induced by a topical teething preparation. Ped Emerg Care. 1987;3:176–178.
11. Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother. 2011;45:1103.
12. Dawson AH, Whyte IM. Management of dapsone poisoning complicated by methemoglobinemia. Med Tox Advers drug Exp. 1989;4:387.
13. Pollack ES, Pollack CV. Incidence of subclinical methemoglobinemia in infants with diarrhea. Ann Emerg Med. 1994;24:652.
14. Nascimento TS, Pereira ROL, Mello HLD, et al. Methemoglobinemia: from diagnosis to treatment. Rev Bras Anestesiol. 2008;58:651.
15. Barker SJ, Badal JJ. The measurement of dyshemoglobins and hemoglobin by pulse oximetry. Curr Opin Anaesthesiol. 2008;21:805.
16. Soeding P, Deppe M, Gehring. Pulse-oximetric measurement of prilocaine-induced methemoglobinemia in regional anesthesia. Anesth Analg. 2010;111:1065.
17. El-Husseini A, Azarov N. Is threshold for treatment of methemoglobinemia the same for all? A case report and literature review. Am J Emerg Med. 2010;28:745.e5.