Nicholas Furtado
HIGH-YIELD FACTS
• Diabetic ketoacidosis (DKA) is a complex endocrine condition caused by an absolute or relative lack of insulin. It is characterized by hyperglycemia, dehydration, ketosis, and metabolic acidosis.
• DKA is often insidious in onset with slow progression of the illness.
• Definition of DKA by biochemical criteria includes the following:
• Hyperglycemia: Blood glucose >200 mg/dL
• Venous pH <7.3 or bicarbonate <15 mmol/L
• Ketonemia and ketonuria
• In type 2 diabetes mellitus, hyperglycemic hyperosmolar state (HHS) can occur and is defined by the following:
• Plasma glucose concentration >600 mg/dL
• Arterial pH >7.30
• Serum bicarbonate >15 mmol/L
• Small ketonuria and absent or mild ketonemia
• Serum osmolarity ≥320 mOsm/kg
• Stupor or coma
Treatment of DKA consists of rapid assessment, replacement of the patient’s fluid and electrolyte deficit, and reversal of the central pathophysiologic process by the administration of insulin.
The initial fluid resuscitation is with normal saline at a dose of 20 mL/kg over 1 to 2 hours. After the initial bolus, the patient’s cardiovascular status is reevaluated and a second bolus may be administered.
An initial bolus of insulin is unnecessary and can increase the risk for cerebral edema. The insulin infusion dose is 0.1 U/kg/h and this should continue till resolution of DKA (pH >7.3, bicarbonate >15 mmol/L).
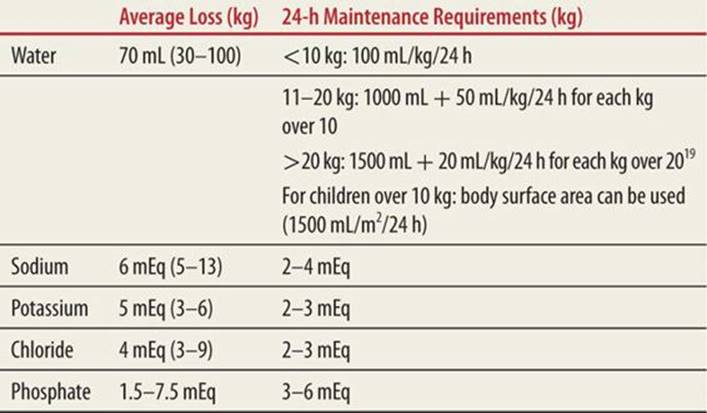
Potassium replacement therapy is started once normal or low serum potassium is ensured and urine output is established. The usual dose of potassium is twice-daily maintenance or 3 to 4 mEq/kg per 24 hours provided as 40 mEq/L in the IV fluids, with half as potassium chloride or potassium acetate and half as potassium phosphate.
Cerebral edema occurs in 0.5% to 0.9% of DKA patients and the mortality rate is 21% to 24%. The predisposing factors are younger age, new onset diabetes, and longer duration of symptoms.
Newborns and young infants with hypoglycemia may be asymptomatic or may manifest nonspecific symptoms. Older children exhibit more classic symptoms of hypoglycemia, including sweating, tachycardia, tremor, anxiety, tachypnea, and weakness.
![]() TREATMENT OF HYPOGLYCEMIA
TREATMENT OF HYPOGLYCEMIA
• In newborns, give 10% dextrose 2 mL/kg (0.2 g/kg) as a bolus, followed by infusion at 6 to 9 mg/kg/min
• In children, give 10% dextrose at 5 mL/kg (0.5 g/kg) as a bolus, followed by continuous infusion at 6 to 9 mg/kg/min
• If an IV line is not possible, then give glucagon 0.03 mg/kg (maximum dose 1 mg) subcutaneously
Admission of the hypoglycemic patient is indicated when there is no obvious cause, toxic ingestion as with oral hypoglycemic agents is suspected, administration of long-acting insulin was the cause, or if there are persistent neurological deficits.
DIABETIC KETOACIDOSIS
DKA is a complex endocrine condition caused by an absolute or relative lack of insulin. It is characterized by hyperglycemia, dehydration, ketosis, and metabolic acidosis.
![]() EPIDEMIOLOGY
EPIDEMIOLOGY
The annual incidence of DKA in the United States ranges from 4.6 to 8 episodes per 1000 patients with diabetes. Diabetes is one of the most common diseases occurring in teenagers. DKA is seen as the initial presentation of diabetes in approximately 25% of young children.1 The risk of DKA in children and adolescents with type 1 diabetes is 1 to 10 per 100 persons per year.2–5 In young patients, DKA accounts for 70% of diabetes-related deaths.
![]() PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
In DKA, a lack of insulin and stress lead to increase in the levels of counterregulatory hormones—glucagon, epinephrine, cortisol, and growth hormone. Gluconeogenesis and glycogenolysis occur in the liver and proteolysis occurs in peripheral tissues. Lipolysis occurs in fatty tissues, forming the ketoacids, α-hydroxybutyrate, and acetoacetic acid. The combination of hyperglycemia and ketoacidosis causes a hyperosmolar diuresis that results in loss of fluids and electrolytes. The combination of ketonemia and hypoperfusion then results in high anion gap metabolic acidosis.
![]() PRECIPITATING FACTORS
PRECIPITATING FACTORS
DKA is precipitated by a variety of causes. DKA at diagnosis is more common in children younger than 5 years and families with poor access to health care.2 In adolescents, noncompliance with insulin is a main cause of DKA. The risk of DKA is increased in peripubertal and adolescent girls, children with clinical depression or eating disorders, and those on insulin pump therapy (due to use of short-acting insulin).2,5,6
![]() CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
History DKA is often insidious in onset with slow progression of the illness. Symptoms can include fatigue and malaise, nausea/vomiting, abdominal pain, polydipsia, polyuria, polyphagia, significant weight loss, and sometimes fever.
Physical Findings Physical findings can include altered mental status characterized by drowsiness, progressive obtundation, and loss of consciousness without evidence of head trauma. The patient will usually have tachycardia, tachypnea or deep, rapid, sighing respirations (Kussmaul respiration), normal or low blood pressure. The child may also have poor perfusion, delayed capillary refill, and other signs of dehydration. The child may have lethargy and weakness, an acetone odor of the breath reflecting metabolic acidosis. Fever may be present if infection precipitated the episode.
Laboratory Studies Initial laboratory studies include a complete blood count, serum electrolytes, glucose, calcium, phosphorus, and serum acetone. The patient may have a nonspecific elevation of serum amylase. An arterial blood gas and bedside tests for blood sugar and urine ketones can be done for rapid diagnosis of DKA. An initial electrocardiogram can be performed to assess for T-wave changes.
Definition of DKA (biochemical criteria)8 is as follows: hyperglycemia with a blood glucose >200 mg/dL; venous pH <7.3 or bicarbonate <15 mmol/L; ketonemia and ketonuria.
DKA can be classified by the degree of acidosis into mild, moderate, and severe.9
• Mild: Venous pH <7.3 or bicarbonate <15 mmol/L
• Moderate: pH <7.2, bicarbonate <10 mmol/L
• Severe: pH <7.1, bicarbonate <5 mmol/L
In type 2 diabetes mellitus, HHS can occur.10 This is defined by the following: plasma glucose concentration >600 mg/dL; arterial pH >7.30; serum bicarbonate >15 mmol/L; small ketonuria and absent or mild ketonemia; serum osmolarity ≥320 mOsm/kg; stupor or coma.
![]() MANAGEMENT
MANAGEMENT
Treatment of DKA consists of rapid assessment, replacement of the patient’s fluid and electrolyte deficit, and reversal of the central pathophysiologic process by the administration of insulin.
Assessment includes performing a quick clinical assessment and bedside tests to confirm the diagnosis. Weigh the patient and use weight for calculation of fluid and electrolyte therapy as well as assess the level of dehydration. Assess the level of consciousness using the Glasgow Coma Scale. Then obtain blood samples and start peripheral IV line and obtain an ECG. Provide supportive measures including airway management for obtunded or comatose patients and oxygen at 100% concentration to patients in respiratory or circulatory failure and shock. Maintain good peripheral or central IV access. Continuous cardiac monitoring is to be used for assessment of T-wave changes 11.
After obtaining samples for cultures, intravenous antibiotics are to be started as soon as possible for patients with DKA precipitated by febrile illness. As soon as hemodynamic stability is achieved, the child should be transferred to an intensive care unit to be managed by a pediatric intensive care specialist with consultation from a pediatric endocrinologist.
![]() FLUID RESUSCITATION
FLUID RESUSCITATION
Children with DKA are at least 5% to 10% dehydrated.12,13 Because clinical estimates are usually inaccurate,14 it is practical to estimate for moderate DKA, 5% to 7% dehydration, and for severe DKA, 7% to 10% dehydration.1 The initial fluid resuscitation is with normal saline at a dose of 20 mL/kg over 1 to 2 hours. After the initial bolus, the patient’s cardiovascular status is reevaluated and a second bolus may be administered. The association between rate of fluid resuscitation and development of cerebral edema is not convincing.15
After the initial fluid resuscitation, rehydration is continued with normal saline or Ringer lactate for 4 to 6 hours depending on state of hydration, serum sodium, and hemodynamic status of the patient.16Subsequently, the remaining fluid deficit should be replaced slowly over 48 hours with a solution of tonicity greater or equal to half normal saline with added potassium chloride, potassium phosphate, or potassium acetate.16–18
In addition to assessment of dehydration, calculation of effective osmolarity can guide fluid and electrolyte therapy. In patients with extreme hyperosmolarity, some recommend continuing therapy with isotonic fluids until serum osmolarity decreases below 320 mOsm/L. The formula for serum osmolality is as follows (blood urea is not included because of low osmolality):
![]()
![]() INSULIN THERAPY
INSULIN THERAPY
The absolute or relative lack of insulin and increase in counterregulatory hormones causes hyperglycemia and DKA. With initial fluid resuscitation, there is some decrease in blood glucose;20,21 however, normalization of blood glucose and suppression of lipolysis requires low-dose, continuous, intravenous insulin infusion.22 This provides slow, reliable, and titratable systemic absorption of insulin. An initial bolus of insulin is unnecessary and can increase the risk for cerebral edema.23,24 The starting dose is 0.1 U/kg/h and this should continue till resolution of DKA (pH >7.3, bicarbonate >15 mmol/L). On occasion, the infusion may have to be decreased to 0.05 U/kg/h when there is marked sensitivity to insulin or conversely it may need to be increased to 0.15 to 0.2 U/kg/h to lower the serum glucose and reverse the ketosis if the patient’s serum glucose is unresponsive to the initial starting dose of 0.1 U/kg/h. The goal of therapy is to decrease the serum glucose by 75 to 100 mg/dL per hour. When the serum glucose reaches 250 mg/dL, 5% glucose is added to the infusing fluid. If the serum glucose is dropping precipitously, a glucose solution of ≥10% may need to be administered. It is dangerous to discontinue the insulin infusion completely if the patient has moderate-to-large serum ketones, since this can worsen the ketoacidosis.
During the initial resuscitation phase, the patient should be given nothing by mouth. As the patient improves, oral intake of water or ice may be provided and advanced to clear liquids as tolerated. When the serum glucose normalizes, metabolic acidosis improves and serum ketones decrease to trace, the insulin infusion is discontinued and switched to subcutaneous insulin and oral intake of liquids or solids. Subcutaneous insulin is administered 30 minutes prior to discontinuing the insulin infusion to allow for absorption of the subcutaneous insulin dose and hence prevent rebound hyperglycemia and ketoacidosis.
If low-dose IV insulin cannot be administered, then subcutaneous (SC) or intramuscular intermittent doses of short- or rapid-acting insulin analog may be used.25 The initial dose is 0.3 U/kg followed in 1 h by SC insulin lispro or as part at 0.1 U/kg every hour or 0.15 to 20 units/kg every 2 hours. If blood sugar falls to below 250 mg/dL before resolution of DKA, then start 5% glucose IV and continue as before. When DKA resolves and blood sugar is <250 mg/dL, reduce insulin to 0.05 U/kg to keep blood sugar at about 200 mg/dL.
![]() POTASSIUM
POTASSIUM
Children with DKA are potassium-depleted with a deficit of 3 to 6 mEq/L/kg.13This loss is primarily intracellular potassium, which is drawn out of the cells by hypertonicity and in exchange for hydrogen ions and also by efflux during glycogenolysis and proteolysis. Potassium is also lost by vomiting and osmotic diuresis.26 Although there is total body depletion of potassium, the initial serum potassium can be normal, increased, or decreased.27 When the insulin infusion is started, potassium is driven back into the cells with decrease in serum levels.28 Hypokalemia is most common after several hours of rehydration. Both severe hypo- and hyperkalemia can cause life-threatening cardiac arrhythmias; therefore, it is essential that the patient’s serum potassium be determined as soon as possible. Alternatively, an ECG can be used to determine if the child has evidence of hyper- or hypokalemia.11 Serum potassium levels should be checked every 2 to 4 hours. Replacement therapy is started once normal or low serum potassium is ensured and urine output is established. The usual dose of potassium is twice-daily maintenance (Table 76-1) or 3 to 4 mEq/kg per 24 hours provided as 40 mEq/L in the IV fluids, with half as potassium chloride or potassium acetate and half as potassium phosphate. The maximum recommended rate of IV potassium is usually 0.5 mEq/kg/h.
|
TABLE 76-1 |
Loss of Fluids and Electrolytes in DKA and Maintenance |
![]() SODIUM
SODIUM
The osmotic diuresis usually induces sodium depletion in patients with DKA. In DKA, both the hyperglycemia and hyperlipidemia cause pseudohyponatremia. In addition, the osmotic movement of water into the extracellular space causes dilutional hyponatremia.29,30 Therefore, corrected serum sodium should be used for monitoring changes during therapy. The formula for corrected serum sodium is as follows:
Corrected serum sodium (mEq/L) = Measured Na + 0.016 × (Serum glucose – 100)
The corrected serum sodium should not be allowed to drop faster than 10 to 12 mEq/L per 24 hours. If significant hyponatremia is present, the first 6 to 8 hours of correction should occur with normal saline. During the continuing resuscitation, sodium levels are monitored every 4 hours and as the glucose falls, the reported level of serum sodium should increase. A fall in serum sodium during continued fluid resuscitation may indicate excess accumulation of free water and may be a risk factor for the development of cerebral edema. If this happens, the sodium content of the fluid may need to be increased.31,32
![]() PHOSPHATE
PHOSPHATE
Depletion of phosphate during DKA occurs as a result of osmotic diuresis.12,13 Clinically significant hypophosphatemia can cause impaired cardiac function and insulin resistance. Usually, after starting insulin, serum phosphate decreases and clinically significant hypophosphatemia can occur if food is not started after 24 hours of fluid therapy.12,13 Prospective studies have not shown clinical benefit from phosphate replacement.33,34 Supplementation is indicated if the serum level is <2 mEq/L and can be administered with potassium replacement as potassium phosphate alone or with potassium chloride. During phosphate replacement, monitor serum calcium for development of hypocalcemia.35,36
![]() ACIDOSIS
ACIDOSIS
The acidosis that is fundamental to DKA is usually reversible with fluid resuscitation and insulin therapy. Controlled trials have shown no clinical benefit from bicarbonate administration.37,38 Bicarbonate causes paradoxical CNS acidosis, rapid onset hypokalemia, and increase in serum osmolality.39,40 Despite these adverse effects and lack of clinical benefit, its cautious use may be considered in patients with severe acidosis (pH <6.9 or serum bicarbonate <5 mEq/L) and hyperkalemia, which are associated with insulin resistance and cardiac arrhythmias. If bicarbonate is considered necessary, administer 1 to 2 mEq/kg over 60 minutes.26
![]() COMPLICATIONS
COMPLICATIONS
Complications of DKA therapy include the following: inadequate rehydration, hypoglycemia, hypokalemia, hyperchloremic acidosis, and cerebral edema. Hypoglycemia is common, especially in young diabetics, who tend to be extremely sensitive to insulin and labile. Adjusting the insulin infusion and providing supplemental intravenous and oral glucose according to the principles outlined above will successfully correct this.
Hypokalemia occurs within several hours of initiation of therapy and can lead to arrhythmias. Therefore, cardiac monitoring is essential during therapy for DKA. Treatment is with potassium replacement, as discussed above.
Cerebral edema occurs in 0.5% to 0.9% of DKA patients and the mortality rate is 21% to 24%.41,42 The predisposing factors are younger age, new onset diabetes, and longer duration of symptoms.43,44 Other risk factors that may be identified at diagnosis or during therapy include administration of insulin in the first hour of fluid replacement, high fluid volume replacement within the first 4 hours,24 severe acidosis, and hypocarbia at presentation after adjusting for acidosis,42 high serum urea nitrogen, attenuated rise of corrected serum sodium during treatment,31 and the use of bicarbonate.42
The warning signs of cerebral edema are as follows: headache and slowing of the heart rate; change in neurological status (restlessness, irritability, increased drowsiness, and incontinence); focal neurological signs (cranial nerve palsy), rising blood pressure; decrease in O2 saturation.
Diagnostic criteria for use in the bedside evaluation of neurological state for the early diagnosis of cerebral edema include the following45:
• Abnormal motor or verbal response to pain
• Decorticate or decerebrate posture
• Cranial nerve palsy (III, IV, and VI)
• Abnormal respiratory pattern (grunting, tachypnea, Cheyne-Stokes breathing, and apnea)
![]() MAJOR CRITERIA
MAJOR CRITERIA
• Altered mentation or fluctuating level of consciousness
• Sustained deceleration of the heart rate
• Age inappropriate incontinence
![]() MINOR CRITERIA
MINOR CRITERIA
• Vomiting
• Headache
• Lethargy (not easily arousable)
• Diastolic BP >90 mm Hg
• Age younger than 5 years
One diagnostic criterion and two major criteria or one major and two minor criteria have a sensitivity of 92% for the diagnosis of cerebral edema in DKA.
![]() TREATMENT OF CEREBRAL EDEMA
TREATMENT OF CEREBRAL EDEMA
Treatment of cerebral edema includes fluid restriction by one-third, the use of IV mannitol 0.5 to 1 g/kg over 20 minutes to be repeated if there is no response in 30 minutes46,47 and hypertonic saline 3%, 5 to 10 mL/kg over 30 minutes.48 Elevate the head of the bed. Consider hyperventilation to maintain a PCO2 <22 mm Hg, although aggressive hyperventilation may be associated with poor outcome.49 After treatment of cerebral edema, a CT scan should be performed to rule out other causes of neurological deterioration such as thrombosis or hemorrhage.
![]() DISPOSITION
DISPOSITION
All patients presenting with DKA as the initial presentation of diabetes are hospitalized at a center where a physician trained in management of pediatric DKA and/or a pediatric endocrinologist are available for consultation. Patients with severe acidosis are best treated in a pediatric intensive care unit for reasons of close monitoring and need for repeated blood sampling.
Children with prolonged illness, decreased level of consciousness, and those at increased risk for cerebral edema at presentation must be admitted to a pediatric intensive care unit for management.8,50Occasionally, children with recurrent and mild DKA, with good family support, may be treated in the emergency department and discharged and followed up as an outpatient in consultation with their endocrinologist.8
HYPOGLYCEMIA
Hypoglycemia is most common in early neonatal life and may reflect a normal adaptive process to extrauterine life.51 In contrast to adults, infants and children are more prone to early development hypoglycemia when fasting because of higher metabolic rate and lower stores of glycogen.52,53 Hypoglycemia is pathological when low blood glucose levels are recurrent and persistent leading to acute systemic effects and long-term neurological sequelae.54,55In childhood and adolescence, hypoglycemia usually presents as a complication of aggressive treatment for insulin-dependent diabetes mellitus56,57(Table 76-2).
|
TABLE 76-2 |
Definition of Hypoglycemia |

The plasma glucose level at which obvious signs and symptoms of hypoglycemia are manifest is variable and depends on the age and clinical characteristics of the patient.
![]() PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
The homeostasis of glucose is maintained by a complex balance between the exogenous supply of food and the body’s regulatory hormones. Insulin and its counterregulatory hormones (glucagon, epinephrine, cortisol, and growth hormone) and their interaction on the liver, muscle, and adipose tissue control glucose levels in the blood. When adequate levels are not maintained, hypoglycemia occurs (Fig. 76-1). Glucose is the main energy substrate for the central nervous system (CNS) and most other organs in the body. Hence, hypoglycemia results in the acute increase in counterregulatory hormones causing autonomic symptoms and CNS dysfunction and sequelae.
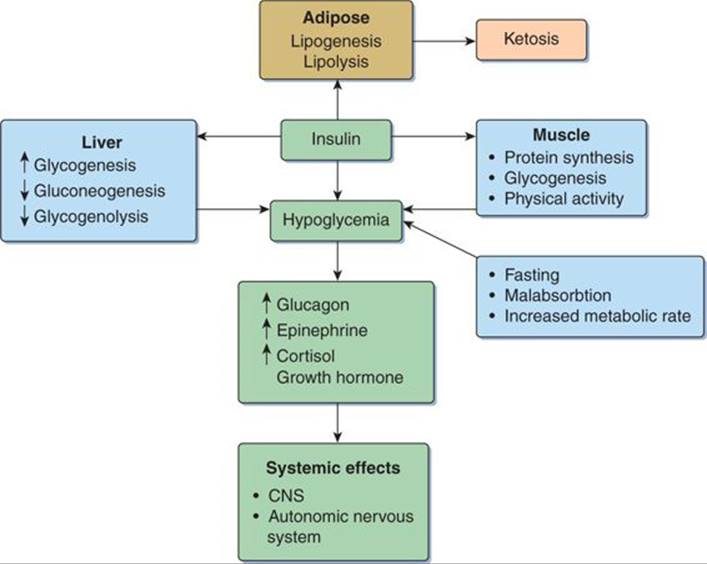
FIGURE 76-1. Pathophysiology of hypoglycemia.
![]() SIGNS AND SYMPTOMS
SIGNS AND SYMPTOMS
Newborns and young infants may be asymptomatic or may manifest nonspecific symptoms (Tables 76-3 and 76-4). Older children exhibit more classic symptoms of hypoglycemia, including sweating, tachycardia, tremor, anxiety, tachypnea, and weakness. Neuroglycopenia, which is a condition of prolonged and severe hypoglycemia manifested on the CNS, can result in permanent neurologic sequelae.
|
TABLE 76-3 |
Symptoms of Hypoglycemia |
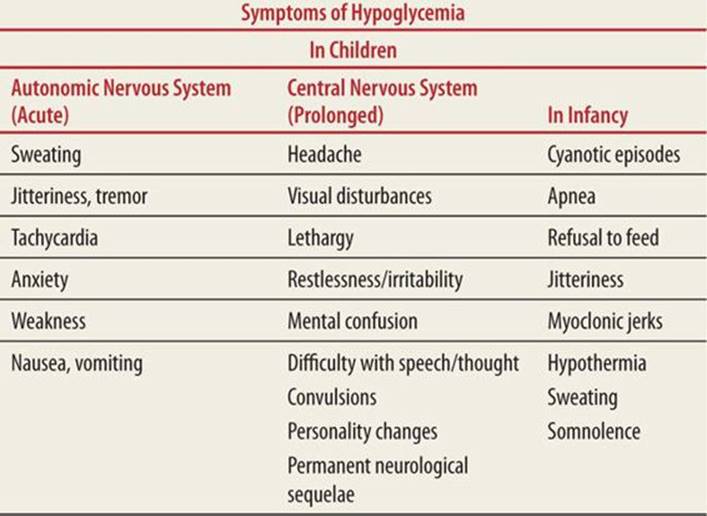
|
TABLE 76-4 |
Etiology of Hypoglycemia in Infants and Children |

![]() DIAGNOSTIC EVALUATIO
DIAGNOSTIC EVALUATIO
Evaluation of an infant or child with hypoglycemia should include a detailed history of the past including perinatal history, acute or recurrent symptoms, and physical examination.58
Ask about perinatal history, including birth weight and maternal diabetes. Ask about dietary relationship to acute symptoms: time after food or association with starvation. Review past history and family history: symptoms, mortality. Also ask parents about growth and development and the possibility of toxic ingestion. Ask about signs and symptoms of the current episode.
Physical examination includes evaluation of anthropometrics (short stature, macrosomia), and for macroglossia, hepatomegaly, jaundice. Evaluate for midline defects (single central incisor, cleft lip/palate, microphallus, and undescended testis). Look for darker skin pigmentation due to adrenal insufficiency. Watch the patient’s respiratory efforts. Is the child hyperventilating (acidosis)?
Laboratory tests include a rapid screen for the plasma glucose level at the bedside. Obtain critical sample which includes repeat blood glucose and other important studies such as insulin, C-peptide, growth hormone, cortisol, and glucagon levels. And obtain a bedside urine test for ketones.
![]() MANAGEMEN
MANAGEMEN
Maintenance of normal plasma glucose levels (70–120 mg/dL) is a must for preserving CNS function (Fig. 76-2).
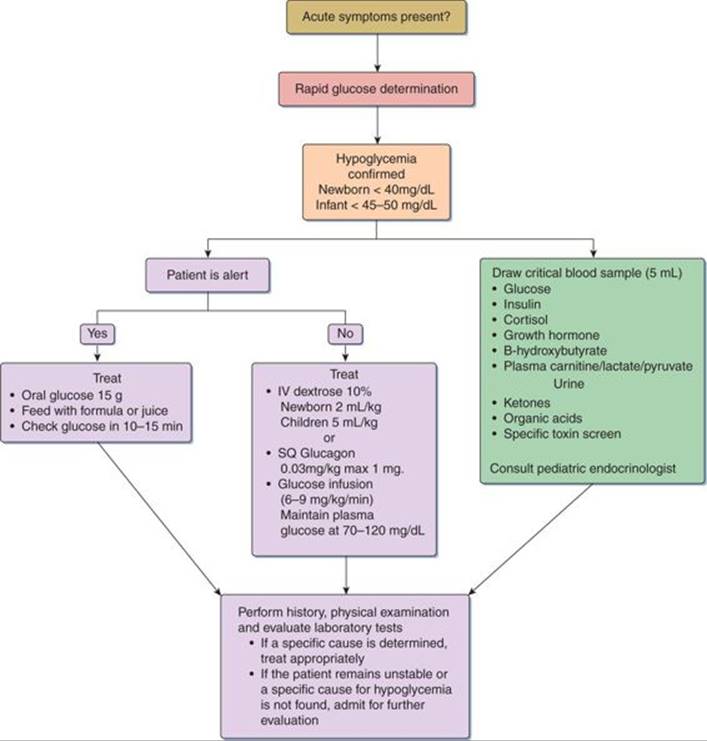
FIGURE 76-2. Acute management of hypoglycemia in infants and children.
![]() PROVIDE ADEQUATE SUBSTRAT
PROVIDE ADEQUATE SUBSTRAT
If the patient is alert, oral glucose 15 g, formula feeds, or juice can be given. If the patient is not alert, start an IV and take the critical blood sample simultaneously. In newborns, give 10% dextrose 2 mL/kg (0.2g/kg) as a bolus, followed by infusion at 6 to 9 mg/kg/min. In children, give 10% dextrose at 5 mL/kg (0.5 g/kg) as a bolus, followed by continuous infusion at 6 to 9 mg/kg/min. If an IV line is not possible, then give glucagon 0.03 mg/kg (maximum dose 1 mg) subcutaneously. If the history, physical examination, and laboratory tests suggest a specific cause for hypoglycemia, then the following drugs may be given as appropriate:
• Growth hormone 0.1 mg/kg/dose
• Hydrocortisone 5 mg/kg/dose po
• Diazoxide 10 to 20 mg/kg/day po
• Somatostatin (Octreotide) 2 to 4 μg/kg/day divided into 2 to 4 doses SQ/IV
• Carnitine 100 mg/kg/day
![]() DISPOSITIO
DISPOSITIO
Admission of the patient is indicated when there is no obvious cause, toxic ingestion as with oral hypoglycemic agents is suspected, administration of long-acting insulin was the cause, and if there are persistent neurological deficits. Discharge may be considered after a high-carbohydrate meal if an obvious cause is found and treated with rapid reversal of symptoms. For all insulin-dependent diabetics with hypoglycemia, discharge should be coordinated with a pediatric endocrinologist and the child’s family after appropriate adjustment of insulin dose.
REFERENCES
1. Wolfsdorf J, Glaser N, Sperling MA. Diabetic ketoacidosis in infants, children, and adolescents: a consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(5):1150–1159.
2. Rewers A, Chase HP, Mackenzie T, et al. Predictors of acute complications in children with type 1 diabetes. JAMA. 2002;287(19):2511–2518.
3. Rosilio M, Cotton JB, Wieliczko MC, et al. Factors associated with glycemic control. A cross-sectional nationwide study in 2,579 French children with type 1 diabetes. The French Pediatric Diabetes Group. Diabetes Care.1998;21(7):1146–1153.
4. Smith CP, Firth D, Bennett S, Howard C, Chisholm P. Ketoacidosis occurring in newly diagnosed and established diabetic children. Acta Paediatr. 1998;87(5):537–541.
5. Hanas R, Lindgren F, Lindblad B. Diabetic ketoacidosis and cerebral oedema in Sweden—a 2-year paediatric population study. Diabet Med. 2007;24(10):1080–1085.
6. Hanas R, Ludvigsson J. Hypoglycemia and ketoacidosis with insulin pump therapy in children and adolescents. Pediatr Diabetes. 2006;7(suppl 4):32–38.
7. Keen H. The diabetes control and complications trial (DCCT). Health Trends. 1994;26(2):41–43.
8. Dunger DB, Sperling MA, Acerini CL, et al. European Society for Paediatric Endocrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics.2004;113(2):e133–e140.
9. Chase HP, Garg SK, Jelley DH. Diabetic ketoacidosis in children and the role of outpatient management. Pediatr Rev. 1990;11(10):297–304.
10. Kitabchi AE, Umpierrez GE, Murphy MB, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care. 2001;24(1):131–153.
11. Malone JI, Brodsky SJ. The value of electrocardiogram monitoring in diabetic ketoacidosis. Diabetes Care. 1980;3(4):543–547.
12. Atchley DW, Loeb RF, Richards DW, Benedict EM, Driscoll ME. On diabetic acidosis: a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest. 1933;12(2):297–326.
13. Nabarro JD, Spencer AG, Stowers JM. Metabolic studies in severe diabetic ketosis. Q J Med. 1952;21(82):225–248.
14. Koves IH, Neutze J, Donath S, et al. The accuracy of clinical assessment of dehydration during diabetic ketoacidosis in childhood. Diabetes Care. 2004;27(10):2485–2487.
15. Brown TB. Cerebral oedema in childhood diabetic ketoacidosis: is treatment a factor? Emerg Med J. 2004;21(2):141–144.
16. Harris GD, Fiordalisi I. Physiologic management of DKA. Arch Dis Child. 2002;87(5):451–452.
17. Hale PM, Rezvani I, Braunstein AW, Lipman TH, Martinez N, Garibaldi L. Factors predicting cerebral edema in young children with diabetic ketoacidosis and new onset type I diabetes. Acta Paediatr.1997;86(6):626–631.
18. Rother KI, Schwenk WF. Effect of rehydration fluid with 75 mmol/L of sodium on serum sodium concentration and serum osmolality in young patients with diabetic ketoacidosis. Mayo Clin Proc.1994;69(12):1149–1153.
19. Friedman AL. Pediatric hydration therapy: historical review and a new approach. Kidney Int. 2005;67(1):380–388.
20. Waldhausl W, Kleinberger G, Korn A, Dudczak R, BratuschMarrain P, Nowotny P. Severe hyperglycemia: effects of rehydration on endocrine derangements and blood glucose concentration. Diabetes.1979;28(6):577–584.
21. Owen OE, Licht JH, Sapir DG. Renal function and effects of partial rehydration during diabetic ketoacidosis. Diabetes. 1981;30(6):510–518.
22. Kitabchi AE. Low-dose insulin therapy in diabetic ketoacidosis: fact or fiction? Diabetes Metab Rev. 1989;5(4):337–363.
23. Dunger DB, Edge JA. Predicting cerebral edema during diabetic ketoacidosis. N Engl J Med. 2001;344(4):302–303.
24. Edge JA, Jakes RW, Roy Y, et al. The UK case-control study of cerebral oedema complicating diabetic ketoacidosis in children. Diabetologia. 2006;49(9):2002–2009.
25. Fisher JN, Shahshahani MN, Kitabchi AE. Diabetic ketoacidosis: low-dose insulin therapy by various routes. N Engl J Med. 1977;297(5):238–241.
26. Wolfsdorf J, Craig ME, Daneman D, et al. Diabetic ketoacidosis. Pediatr Diabetes. 2007;8(1):28–43.
27. Adrogue HJ, Lederer ED, Suki WN, Eknoyan G. Determinants of plasma potassium levels in diabetic ketoacidosis. Medicine (Baltimore). 1986;65(3):163–172.
28. DeFronzo RA, Felig P, Ferrannini E, Wahren J. Effect of graded doses of insulin on splanchnic and peripheral potassium metabolism in man. Am J Physiol. 1980;238(5):E421–E427.
29. Katz MA. Hyperglycemia-induced hyponatremia—calculation of expected serum sodium depression. N Engl J Med. 1973;289(16):843–844.
30. Hillier TA, Abbott RD, Barrett EJ. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med. 1999;106(4):399–403.
31. Harris GD, Fiordalisi I, Harris WL, Mosovich LL, Finberg L. Minimizing the risk of brain herniation during treatment of diabetic ketoacidemia: a retrospective and prospective study. J Pediatr. 1990;117(1 Pt 1):22–31.
32. Duck SC, Wyatt DT. Factors associated with brain herniation in the treatment of diabetic ketoacidosis. J Pediatr. 1988;113(1 Pt 1):10–14.
33. Gibby OM, Veale KE, Hayes TM, Jones JG, Wardrop CA. Oxygen availability from the blood and the effect of phosphate replacement on erythrocyte 2,3-diphosphoglycerate and haemoglobin-oxygen affinity in diabetic ketoacidosis. Diabetologia. 1978;15(5):381–385.
34. Wilson HK, Keuer SP, Lea AS, Boyd AE III, Eknoyan G. Phosphate therapy in diabetic ketoacidosis. Arch Intern Med. 1982;142(3):517–520.
35. Zipf WB, Bacon GE, Spencer ML, Kelch RP, Hopwood NJ, Hawker CD. Hypocalcemia, hypomagnesemia, and transient hypoparathyroidism during therapy with potassium phosphate in diabetic ketoacidosis. Diabetes Care.1979;2(3):265–268.
36. Winter RJ, Harris CJ, Phillips LS, Green OC. Diabetic ketoacidosis. Induction of hypocalcemia and hypomagnesemia by phosphate therapy. Am J Med. 1979;67(5):897–900.
37. Hale PJ, Crase J, Nattrass M. Metabolic effects of bicarbonate in the treatment of diabetic ketoacidosis. Br Med J (Clin Res Ed). 1984;289(6451):1035–1038.
38. Green SM, Rothrock SG, Ho JD, et al. Failure of adjunctive bicarbonate to improve outcome in severe pediatric diabetic ketoacidosis. Ann Emerg Med. 1998;31(1):41–48.
39. Assal JP, Aoki TT, Manzano FM, Kozak GP. Metabolic effects of sodium bicarbonate in management of diabetic ketoacidosis. Diabetes. 1974;23(5):405–411.
40. Soler NG, Bennett MA, Dixon K, FitzGerald MG, Malins JM. Potassium balance during treatment of diabetic ketoacidosis with special reference to the use of bicarbonate. Lancet. 1972;2(7779):665–667.
41. Edge JA, Hawkins MM, Winter DL, Dunger DB. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85(1):16–22.
42. Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001;344(4):264–269.
43. Rosenbloom AL. Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes Care. 1990;13(1):22–33.
44. Bello FA, Sotos JF. Cerebral oedema in diabetic ketoacidosis in children. Lancet. 1990;336(8706):64.
45. Muir AB, Quisling RG, Yang MC, Rosenbloom AL. Cerebral edema in childhood diabetic ketoacidosis: natural history, radiographic findings, and early identification. Diabetes Care. 2004;27(7):1541–1546.
46. Franklin B, Liu J, Ginsberg-Fellner F. Cerebral edema and ophthalmoplegia reversed by mannitol in a new case of insulin-dependent diabetes mellitus. Pediatrics 1982;69(1):87–90.
47. Shabbir N, Oberfield SE, Corrales R, Kairam R, Levine LS. Recovery from symptomatic brain swelling in diabetic ketoacidosis. Clin Pediatr (Phila). 1992;31(9):570–573.
48. Curtis JR, Bohn D, Daneman D. Use of hypertonic saline in the treatment of cerebral edema in diabetic ketoacidosis (DKA). Pediatr Diabetes. 2001;2(4):191–194.
49. Marcin JP, Glaser N, Barnett P, et al. Factors associated with adverse outcomes in children with diabetic ketoacidosis-related cerebral edema. J Pediatr. 2002;141(6):793–797.
50. Monroe KW, King W, Atchison JA. Use of PRISM scores in triage of pediatric patients with diabetic ketoacidosis. Am J Manag Care. 1997;3(2):253–258.
51. Cornblath M. Neonatal hypoglycemia 30 years later: does it injure the brain? Historical summary and present challenges. Acta Paediatr Jpn. 1997;39(suppl 1):S7–S11.
52. Haymond MW, Karl IE, Clarke WL, Pagliara AS, Santiago JV. Differences in circulating gluconeogenic substrates during short-term fasting in men, women, and children. Metabolism. 1982;31(1):33–42.
53. Chaussain JL, Georges P, Calzada L, Job JC. Glycemic response to 24-hour fast in normal children: III. Influence of age. J Pediatr. 1977;91(5):711–714.
54. Pildes RS, Cornblath M, Warren I, et al. A prospective controlled study of neonatal hypoglycemia. Pediatrics. 1974;54(1):5–14.
55. Anderson JM, Milner RD, Strich SJ. Effects of neonatal hypoglycaemia on the nervous system: a pathological study. J Neurol Neurosurg Psychiatry. 1967;30(4):295–310.
56. Becker DJ, Ryan CM. Hypoglycemia: a complication of diabetes therapy in children. Trends Endocrinol Metab. 2000;11(5):198–202.
57. Ryan C, Gurtunca N, Becker D. Hypoglycemia: a complication of diabetes therapy in children. Pediatr Clin North Am. 2005;52(6):1705–1733.
58. Haymond MW. Hypoglycemia in infants and children. Endocrinol Metab Clin North Am. 1989;18(1):211–252.