Ian A. Reid, PhD
CASE STUDY
During a routine check, a 45-year-old man was found to have high blood pressure (165/100 mm Hg). Blood pressure remained high on two follow-up visits. His physician initially prescribed hydrochlorothiazide, a diuretic commonly used to treat hypertension. Although his blood pressure was reduced by hydrochlorothiazide, it remained at a hypertensive level (145/95 mm Hg), and he was referred to the university hypertension clinic. Your evaluation revealed that the patient had elevated plasma renin activity and aldosterone concentration. Hydrochlorothiazide was therefore replaced with enalapril, an angiotensin-converting enzyme inhibitor. Enalapril lowered the blood pressure to almost normotensive levels. However, after several weeks on enalapril, the patient now returns complaining of a persistent cough. In addition, some signs of angioedema are detected. How does enalapril lower blood pressure? Why does it occasionally cause coughing and angioedema? What other drugs could be used to inhibit renin secretion or suppress the renin-angiotensin system, and decrease blood pressure, without the adverse effects of enalapril?
Peptides are used by most tissues for cell-to-cell communication. As noted in Chapters 6 and 21, they play important roles as transmitters in the autonomic and central nervous systems. Several peptides exert important direct effects on vascular and other smooth muscles. These peptides include vasoconstrictors (angiotensin II, vasopressin, endothelins, neuropeptide Y, and urotensin) and vasodilators (bradykinin and related kinins, natriuretic peptides, vasoactive intestinal peptide, substance P, neurotensin, calcitonin gene-related peptide, and adrenomedullin). This chapter focuses on the smooth muscle actions of the peptides and on drugs that alter their biosynthesis or actions.
![]() ANGIOTENSIN
ANGIOTENSIN
BIOSYNTHESIS OF ANGIOTENSIN
The pathway for the formation and metabolism of angiotensin II (ANG II) is summarized in Figure 17–1. The principal steps include enzymatic cleavage of angiotensin I (ANG I) from angiotensinogen by renin, conversion of ANG I to ANG II by converting enzyme, and degradation of ANG II by several peptidases.
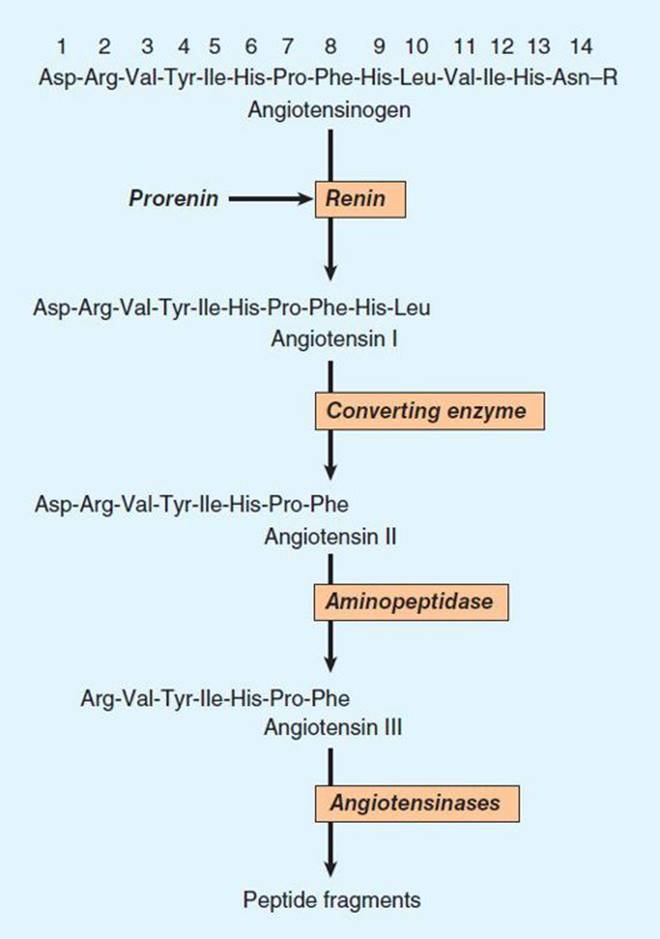
FIGURE 17–1 Chemistry of the renin-angiotensin system. The amino acid sequence of the amino terminal of human angiotensinogen is shown. R denotes the remainder of the protein molecule. See text for additional steps in the formation and metabolism of angiotensin peptides.
Renin
Renin is an aspartyl protease enzyme that specifically catalyzes the hydrolytic release of the decapeptide ANG I from angiotensinogen. It is synthesized as a prepromolecule that is processed to prorenin, which has poorly understood actions, and then to active renin, a glycoprotein consisting of 340 amino acids.
Renin in the circulation originates in the kidneys. Enzymes with renin-like activity are present in several extrarenal tissues, including blood vessels, uterus, salivary glands, and adrenal cortex, but no physiologic role for these enzymes has been established. Within the kidney, renin is synthesized and stored in the juxtaglomerular apparatus of the nephron. Specialized granular cells called juxtaglomerular cells are the site of synthesis, storage, and release of renin. The macula densa is a specialized segment of the nephron that is closely associated with the vascular components of the juxtaglomerular apparatus. The vascular and tubular components of the juxtaglomerular apparatus, including the juxtaglomerular cells, are innervated by the sympathetic nervous system.
Control of Renin Release
The rate at which renin is released by the kidney is the primary determinant of activity of the renin-angiotensin system. Active renin is released by exocytosis immediately upon stimulation of the juxtaglomerular apparatus. Prorenin is released constitutively, usually at a rate higher than that of active renin, thus accounting for the fact that prorenin can constitute 80–90% of the total renin in the circulation. The significance of circulating prorenin is discussed at the end of this section. Active renin release is controlled by a variety of factors, including the macula densa, a renal vascular receptor, the sympathetic nervous system, and ANG II.
A. Macula Densa
Renin release is controlled in part by the macula densa, a structure that has a close anatomic association with the afferent arteriole. The initial step involves the detection of some function of NaCl concentration in, or delivery to, the distal tubule, possibly by the Na+/K+/2Cl− cotransporter. The macula densa then signals changes in renin release by the juxtaglomerular cells such that there is an inverse relationship between NaCl delivery or concentration and renin release. Potential candidates for signal transmission include prostaglandin E2 (PGE2) and nitric oxide, which stimulate renin release, and adenosine, which inhibits it. Because the sodium intake in the general population is high, macula densa-mediated renin secretion is usually at basal levels, increasing only when sodium intake decreases.
B. Renal Baroreceptor
The renal vascular baroreceptor mediates an inverse relationship between renal artery pressure and renin release. The mechanism is not completely understood but it appears that the juxtaglomerular cells are sensitive to stretch and that increased stretch results in decreased renin release. The decrease may result from influx of calcium which, somewhat paradoxically, inhibits renin release. The paracrine factors PGE2, nitric oxide, and adenosine have also been implicated in the baroreceptor control of renin release. At normal blood pressure, renal baroreceptor-mediated renin secretion is low; it increases in hypotensive states.
C. Sympathetic Nervous System
Norepinephrine released from renal sympathetic nerves stimulates renin release indirectly by α-adrenergic activation of the renal baroreceptor and macula densa mechanisms, and directly by an action on the juxtaglomerular cells. In humans, the direct effect is mediated by β1 adrenoceptors. Through this mechanism, reflex activation of the sympathetic nervous system by hypotension or hypovolemia leads to activation of the renin-angiotensin system.
D. Angiotensin
Angiotensin II inhibits renin release. The inhibition results from increased blood pressure acting by way of the renal baroreceptor and macula densa mechanisms, and from a direct action of the peptide on the juxtaglomerular cells. The direct inhibition is mediated by increased intracellular Ca2+ concentration and forms the basis of a short-loop negative feedback mechanism controlling renin release. Interruption of this feedback with drugs that inhibit the renin-angiotensin system results in stimulation of renin release.
E. Intracellular Signaling Pathways
The release of renin by the juxtaglomerular cells is controlled by interplay among three intracellular messengers: cAMP, cyclic guanosine monophosphate (cGMP), and free cytosolic Ca2+ concentration (Figure 17–2). cAMP plays a major role; maneuvers that increase cAMP levels, including activation of adenylyl cyclase, inhibition of cAMP phosphodiesterases, and administration of cAMP analogs, increase renin release. In experimental studies, selective deficiency of Gsα in the juxtaglomerular cells is associated with a marked reduction in basal renin secretion and in the response to several stimuli to renin secretion.
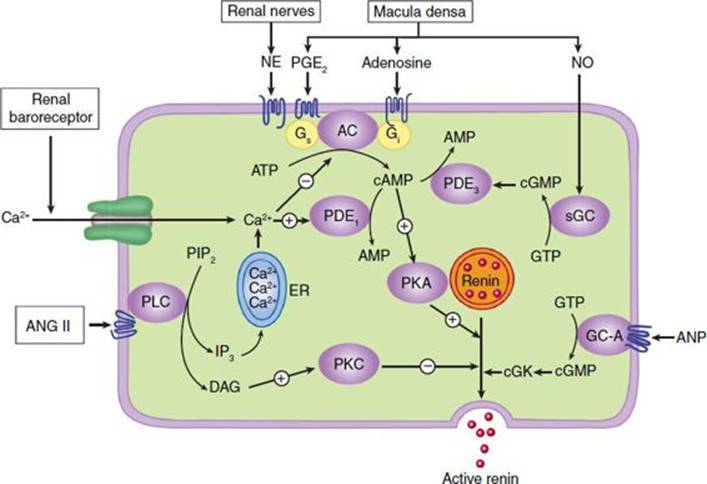
FIGURE 17–2 Major physiologic inputs to renin release and proposed integration with signaling pathways in the juxtaglomerular cell. AC, adenylyl cyclase; ANG II, angiotensin II; ANP, atrial natriuretic peptide; cGK, protein kinase G; DAG, diacylglycerol; GC-A, particulate guanylyl cyclase; ER, endoplasmic reticulum; IP3, inositol trisphosphate; NE, norepinephrine; NO, nitric oxide; PDE, phosphodiesterase; PKA, protein kinase A; PLC, phospholipase C; sGC, soluble guanylyl cyclase. (Adapted, with permission, from Castrop H et al: Physiology of kidney renin. Physiol Rev 2010;90:607.)
Increases in intracellular Ca2+ can result from increased entry of extracellular Ca2+ or mobilization of Ca2+ from intracellular stores, while increases in cGMP levels can result from activation of soluble or particulate guanylyl cyclase. Ca2+ and cGMP appear to alter renin release indirectly, primarily by changing cAMP levels.
F. Pharmacologic Alteration of Renin Release
The release of renin is altered by a wide variety of pharmacologic agents. Renin release is stimulated by vasodilators (hydralazine, minoxidil, nitroprusside), β-adrenoceptor agonists, α-adrenoceptor antagonists, phosphodiesterase inhibitors (eg, theophylline, milrinone, rolipram), and most diuretics and anesthetics. This stimulation can be accounted for by the control mechanisms just described. Drugs that inhibit renin release are discussed below.
Many of the peptides reviewed in this chapter also alter renin release. Release is stimulated by adrenomedullin, bradykinin, and calcitonin gene-related peptide, and inhibited by atrial natriuretic peptide, endothelin, substance P, and vasopressin.
Angiotensinogen
Angiotensinogen is the circulating protein substrate from which renin cleaves ANG I. It is synthesized in the liver. Human angiotensinogen is a glycoprotein with a molecular weight of approximately 57,000. The 14 amino acids at the amino terminal of the molecule are shown in Figure 17–1. In humans, the concentration of angiotensinogen in the circulation is less than the Km of the renin-angiotensinogen reaction and is therefore an important determinant of the rate of formation of angiotensin.
The production of angiotensinogen is increased by corticosteroids, estrogens, thyroid hormones, and ANG II. It is elevated during pregnancy and in women taking estrogen-containing oral contraceptives. The increased plasma angiotensinogen concentration is thought to contribute to the hypertension that may occur in these situations.
Angiotensin I
Although ANG I contains the peptide sequences necessary for all of the actions of the renin-angiotensin system, it has little or no biologic activity. Instead, it must be converted to ANG II by converting enzyme (Figure 17–1). ANG I may also be acted on by plasma or tissue aminopeptidases to form [des-Asp1]angiotensin I; this in turn is converted to [des-Asp1]angiotensin II (commonly known as angiotensin III) by converting enzyme.
Converting Enzyme (ACE, Peptidyl Dipeptidase, Kininase II)
Converting enzyme is a dipeptidyl carboxypeptidase with two active sites that catalyzes the cleavage of dipeptides from the carboxyl terminal of certain peptides. Its most important substrates are ANG I, which it converts to ANG II, and bradykinin, which it inactivates (see Kinins, below). It also cleaves enkephalins and substance P, but the physiologic significance of these effects has not been established. The action of converting enzyme is prevented by a penultimate prolyl residue in the substrate, and ANG II is therefore not hydrolyzed by converting enzyme. Converting enzyme is distributed widely in the body. In most organs, converting enzyme is located on the luminal surface of vascular endothelial cells and is thus in close contact with the circulation.
A homolog of converting enzyme known as ACE2 was recently found to be highly expressed in vascular endothelial cells of the kidneys, heart, and testes. Unlike converting enzyme, ACE2 has only one active site and functions as a carboxypeptidase rather than a dipeptidyl carboxypeptidase. It removes a single amino acid from the C-terminal of ANG I forming ANG 1-9 (Figure 17–3), which is inactive but is converted to ANG 1-7 by ACE. ACE2 also converts ANG II to ANG 1-7. ANG 1-7 has vasodilator activity, apparently mediated by the orphan heterotrimeric guanine nucleotide-binding protein-coupled receptor (Mas receptor). This vasodilation may serve to counteract the vasoconstrictor activity of ANG II. ACE2 also differs from ACE in that it does not hydrolyze bradykinin and is not inhibited by converting enzyme inhibitors (see below). Thus, ACE2 more closely resembles an angiotensinase than a converting enzyme.
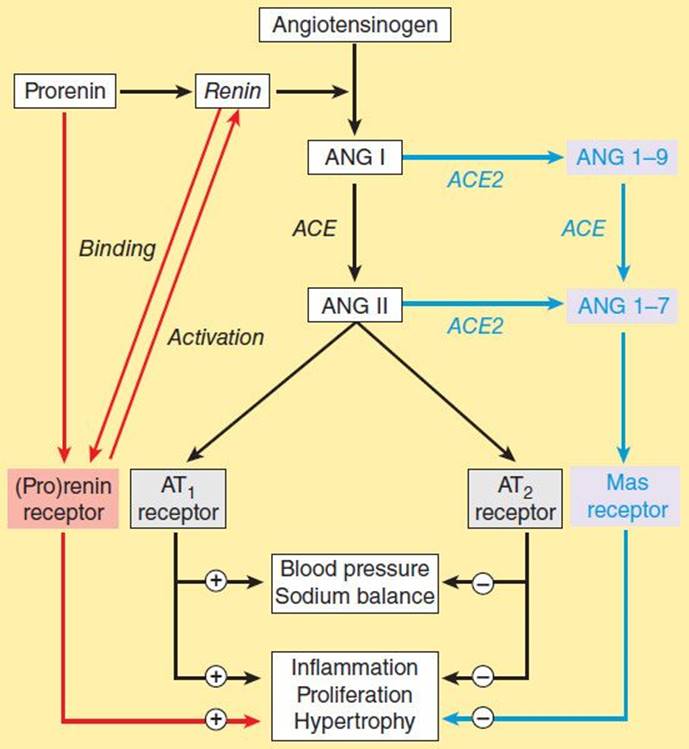
FIGURE 17–3 The renin-angiotensin system showing the established system (black) and recently discovered pathways involving the (pro)renin receptor (red) and ANG 1-7 (blue). (Adapted, with permission, from Castrop H et al: Physiology of kidney renin. Physiol Rev 2010;90:607.)
Angiotensinase
Angiotensin II, which has a plasma half-life of 15–60 seconds, is removed rapidly from the circulation by a variety of peptidases collectively referred to as angiotensinase. It is metabolized during passage through most vascular beds (a notable exception being the lung). Most metabolites of ANG II are biologically inactive, but the initial product of aminopeptidase action—[des-Asp1]angiotensin II—retains considerable biologic activity.
ACTIONS OF ANGIOTENSIN II
Angiotensin II exerts important actions at vascular smooth muscle, adrenal cortex, kidney, heart, and brain via the receptors described below. Through these actions, the renin-angiotensin system plays a key role in the regulation of fluid and electrolyte balance and arterial blood pressure. Excessive activity of the renin-angiotensin system can result in hypertension and disorders of fluid and electrolyte homeostasis.
Blood Pressure
Angiotensin II is a potent pressor agent—on a molar basis, approximately 40 times more potent than norepinephrine. The pressor response to intravenous ANG II is rapid in onset (10–15 seconds) and sustained during long-term infusions. A large component of the pressor response is due to direct contraction of vascular—especially arteriolar—smooth muscle. In addition, however, ANG II can also increase blood pressure through actions on the brain and autonomic nervous system. The pressor response to ANG II is usually accompanied by little or no reflex bradycardia because the peptide simultaneously acts on the brain to reset the baroreceptor reflex control of heart rate to a higher pressure.
Angiotensin II also interacts with the autonomic nervous system. It stimulates autonomic ganglia, increases the release of epinephrine and norepinephrine from the adrenal medulla, and most important, facilitates sympathetic transmission by an action at adrenergic nerve terminals. The latter effect involves both increased release and reduced reuptake of norepinephrine. Angiotensin II also has a less important direct positive inotropic action on the heart.
Adrenal Cortex & Kidney
Angiotensin II acts directly on the zona glomerulosa of the adrenal cortex to stimulate aldosterone synthesis and release. At higher concentrations, ANG II also stimulates glucocorticoid synthesis. Angiotensin II acts on the kidney to cause renal vasoconstriction, increase proximal tubular sodium reabsorption, and inhibit the release of renin.
Central Nervous System
In addition to its central effects on blood pressure, ANG II acts on the central nervous system to stimulate drinking (dipsogenic effect) and increase the secretion of vasopressin and adrenocorticotropic hormone (ACTH). The physiologic significance of these effects is not known.
Cell Growth
Angiotensin II is mitogenic for vascular and cardiac muscle cells and may contribute to the development of cardiovascular hypertrophy. It also exerts a variety of important effects on the vascular endothelium. Indeed, overactivity of the renin-angiotensin system has been implicated as one of the most significant factors in the development of hypertensive vascular disease. Considerable evidence now indicates that ACE inhibitors and ANG II receptor antagonists (see below) slow or prevent morphologic changes (remodeling) following myocardial infarction that would otherwise lead to heart failure. The stimulation of vascular and cardiac growth by ANG II is mediated by other pathways, probably receptor and nonreceptor tyrosine kinases such as the Janus tyrosine kinase Jak2, and by increased transcription of specific genes (see Chapter 2).
ANGIOTENSIN RECEPTORS & MECHANISM OF ACTION
Angiotensin II receptors are widely distributed in the body. Like the receptors for other peptide hormones, ANG II receptors are G protein-coupled and located on the plasma membrane of target cells, and this permits rapid onset of the various actions of ANG II. Two distinct subtypes of ANG II receptors, termed AT1 and AT2, have been identified on the basis of their differential affinity for antagonists and their sensitivity to sulfhydryl-reducing agents. AT1receptors have a high affinity for the inhibitor losartan and a low affinity for PD 123177 (an experimental nonpeptide antagonist), whereas AT2 receptors have a high affinity for PD 123177 and a low affinity for losartan. Angiotensin II binds equally to both subtypes. The relative proportion of the two subtypes varies from tissue to tissue: AT1 receptors predominate in vascular smooth muscle. Most of the known actions of ANG II are mediated by the AT1receptor, a Gq protein-coupled receptor. Binding of ANG II to AT1 receptors in vascular smooth muscle results in activation of phospholipase C and generation of inositol trisphosphate and diacylglycerol (see Chapter 2). These events, which occur within seconds, result in smooth muscle contraction.
The AT2 receptor has a structure and affinity for ANG II similar to those of the AT1 receptor. In contrast, however, stimulation of AT2 receptors causes vasodilation that may serve to counteract the vasoconstriction resulting from AT1 receptor stimulation. AT2 receptor-mediated vasodilation appears to be nitric oxide-dependent and may involve the bradykinin B2 receptor-nitric oxide-cGMP pathway. AT2receptors are present at high density in all tissues during fetal development, but they are much less abundant in the adult where they are expressed at high concentration only in the adrenal medulla, reproductive tissues, vascular endothelium, and parts of the brain. AT2 receptors are up-regulated in pathologic conditions including heart failure and myocardial infarction. The functions of the AT2 receptor appear to include fetal tissue development, inhibition of growth and proliferation, cell differentiation, apoptosis, and vasodilation.
INHIBITION OF THE RENIN-ANGIOTENSIN SYSTEM
In view of the importance of the renin-angiotensin system in cardiovascular disease, considerable effort has been directed to developing drugs that inhibit the system. A wide variety of agents that block the formation or action of ANG II is now available. Some of these drugs block renin release, but most inhibit the conversion of ANG I to ANG II, block angiotensin AT1 receptors, or inhibit the enzymatic action of renin.
Drugs that Block Renin Release
Drugs that interfere with the sympathetic nervous system inhibit the release of renin. Examples are propranolol and other β-adrenoceptor-blocking drugs, which act by blocking the renal β receptors involved in the sympathetic control of renin release.
Angiotensin-Converting Enzyme Inhibitors
An important class of orally active ACE inhibitors, directed against the active site of ACE, is now extensively used. Captopril and enalapril are examples of the many ACE inhibitors that are available. These drugs differ in their structure and pharmacokinetics, but they are interchangeable in clinical use. ACE inhibitors decrease systemic vascular resistance without increasing heart rate, and they promote natriuresis. As described in Chapters 11 and 13, they are effective in the treatment of hypertension, decrease morbidity and mortality in heart failure and left ventricular dysfunction after myocardial infarction, and delay the progression of diabetic nephropathy.
ACE inhibitors not only block the conversion of ANG I to ANG II but also inhibit the degradation of other substances, including bradykinin, substance P, and enkephalins. The action of ACE inhibitors to inhibit bradykinin metabolism contributes significantly to their hypotensive action (see Figure 11–5) and is apparently responsible for some adverse side effects, including cough and angioedema. These drugs are contraindicated in pregnancy because they cause fetal kidney damage.
Angiotensin Receptor Blockers
Peptide antagonists of the action of ANG II are available for research use. However, the nonpeptide ANG II receptor blockers (ARBs) are of much greater interest. Losartan, valsartan, and several others are orally active, potent, and specific competitive antagonists of angiotensin AT1 receptors. The efficacy of these drugs in hypertension is similar to that of ACE inhibitors, but they are associated with a lower incidence of cough. Like ACE inhibitors, ARBs slow the progression of diabetic nephropathy and valsartan has been reported to decrease the incidence of diabetes in patients with impaired glucose tolerance. The antagonists are also effective in the treatment of heart failure and provide a useful alternative when ACE inhibitors are not well tolerated. ARBs are generally well tolerated but should not be used by patients with nondiabetic renal disease or in pregnancy. In addition, some ARBs may cause a syndrome known as sprue-like enteropathy.
Marfan syndrome is a connective tissue disorder associated with aortic disease and other abnormalities involving increased transforming growth factor (TGF)-β signaling. Since ANG II increases TGF-β levels, it was reasoned that blockade of the renin-angiotensin system might be beneficial in Marfan syndrome. Promising initial results have been obtained with losartan, and clinical trials are underway.
The currently available ARBs are selective for the AT1 receptor. Since prolonged treatment with the drugs disinhibits renin release and increases circulating ANG II levels, there may be increased stimulation of AT2 receptors. This may be significant in view of the evidence that activation of the AT2 receptor causes vasodilation and other beneficial effects. AT2 receptor antagonists such as PD 123177 are available for research but have no clinical applications at this time. However, a selective AT2 agonist, compound 21, lowers blood pressure in hypertensive animals and may be beneficial in human hypertension. The clinical benefits of ARBs are similar to those of ACE inhibitors, and it is not clear if either group has significant advantages over the other.
Renin Inhibitors
Cleavage of angiotensinogen by renin (Figures 17–1 and 17–3) is the rate-limiting step in the formation of ANG II and thus represents a logical target for inhibition of the renin-angiotensin system. Drugs that inhibit renin have been available for many years but have been limited by low potency, poor bioavailability, and short duration of action. However, a new class of nonpeptide, low-molecular-weight, orally active inhibitors is now available.
Aliskiren is the first nonpeptide renin inhibitor to be approved for the treatment of hypertension. In healthy subjects, aliskiren produces dose-dependent reductions in plasma renin activity and ANG I and II and aldosterone concentrations. In patients with hypertension, many of whom have elevated plasma renin levels, aliskiren suppresses plasma renin activity and causes dose-related reductions in blood pressure similar to those produced by ACE inhibitors and ARBs. The safety and tolerability of aliskiren appear to be comparable to angiotensin antagonists and placebo. Thus, renin inhibition may be an important approach to the treatment of hypertension. Aliskiren is contraindicated in pregnancy.
Inhibition of the renin-angiotensin system with ACE inhibitors or ARBs may be incomplete because the drugs disrupt the negative feedback action of ANG II on renin release and thereby increase plasma renin activity. Other antihypertensive drugs, notably hydrochlorothiazide and other diuretics, also increase plasma renin activity. Aliskiren not only decreases baseline plasma renin activity in hypertensive subjects but also eliminates the rise produced by ACE inhibitors, ARBs, and diuretics, and thereby results in a greater antihypertensive effect. For this reason, aliskiren has been used in combination with an ACE inhibitor or ARB. However, such dual blockade may not produce significant clinical benefit and may even be associated with adverse effects including hypokalemia, although this point is controversial.
Prorenin Receptors
For many years, prorenin was considered to be an inactive precursor of renin, with no function of its own. Thus the observation noted above in the section on renin that prorenin circulates at high levels was surprising. Recently, however, a receptor that preferentially binds prorenin has been identified. Since it also binds active renin, the receptor is referred to as the (pro)renin receptor.
The receptor is a 350-amino acid protein with a single transmembrane domain. When prorenin binds to the (pro)renin receptor, the prorenin undergoes a conformational change and becomes enzymatically active without cleavage of the prosegment. The catalytic activity of active renin also increases further when it binds to the receptor. The activated prorenin and renin interact with circulating angiotensinogen to form angiotensin (Figure 17–3). However, binding of prorenin to the receptor also activates intracellular signaling pathways that differ depending on the cell type. For example, in mesangial and vascular smooth muscle cells, prorenin binding activates MAP kinases and expression of profibrotic molecules. Thus, elevated prorenin levels (as occur, for example, in diabetes mellitus) could produce a variety of adverse effects via both angiotensin-dependent and independent pathways. Recent research indicates that the (pro)renin receptor is functionally linked to the vacuolar proton-ATPase (ATP6ap2) and is necessary for Wnt signaling pathways involved (independently of renin) in stem cell biology, embryology, and cancer.
The renin activity of the receptor-bound forms of prorenin and renin can be inhibited by aliskiren. A synthetic peptide named handle region peptide (HRP), which consists of the amino acid sequence corresponding to the “handle” region of the prorenin prosegment, has been synthesized and shown to competitively inhibit binding of prorenin to the (pro)renin receptor. HRP has beneficial effects in the kidneys of diabetic rats and so there is considerable interest in developing noncompetitive antagonists of the (pro)renin receptor.
This novel receptor could be important in cardiovascular and other diseases, but at the present time its role in human pathology is far from clear.
![]() KININS
KININS
BIOSYNTHESIS OF KININS
Kinins are potent vasodilator peptides formed enzymatically by the action of enzymes known as kallikreins acting on protein substrates called kininogens. The kallikrein-kinin system has several features in common with the renin-angiotensin system.
Kallikreins
Kallikreins are serine proteases present in plasma (plasma kallikrein) and in several organs (tissue kallikrein), including the kidneys, pancreas, intestine, sweat glands, and salivary glands. The two groups are secreted as zymogens and are activated by proteolytic cleavage. Plasma prekallikrein is activated by activated blood coagulation factor XII (FXIIa). The two groups differ in their gene structure, molecular weight, substrate specificity, and kinin produced. Kallikreins can convert prorenin to active renin, but the physiologic significance of this action is not known.
Kininogens
Kininogens—the substrates for kallikreins and precursors of kinins—are present in plasma, lymph, and interstitial fluid. Two kininogens are present in plasma: a low-molecular-weight form (LMW kininogen) and a high-molecular-weight form (HMW kininogen). The two forms result from differential splicing of the kininogen gene to generate proteins that differ at the C-terminus. About 15–20% of the total plasma kininogen is in the HMW form. It is thought that LMW kininogen crosses capillary walls and serves as the substrate for tissue kallikreins, whereas HMW kininogen is confined to the bloodstream and serves as the substrate for plasma kallikrein.
FORMATION & METABOLISM OF KININS
The pathway for the formation and metabolism of kinins is shown in Figure 17–4. The two major kinins in humans are bradykinin and Lys-bradykinin or kallidin. Bradykinin is released from HMW kininogen by plasma kallikrein, whereas kallidin is released from LMW kininogen by tissue kallikrein. Kallidin can be converted to bradykinin by an arginine aminopeptidase. The two kinins are present in plasma and urine. Bradykinin is the predominant kinin in plasma, whereas Lys-bradykinin is the major urinary form.
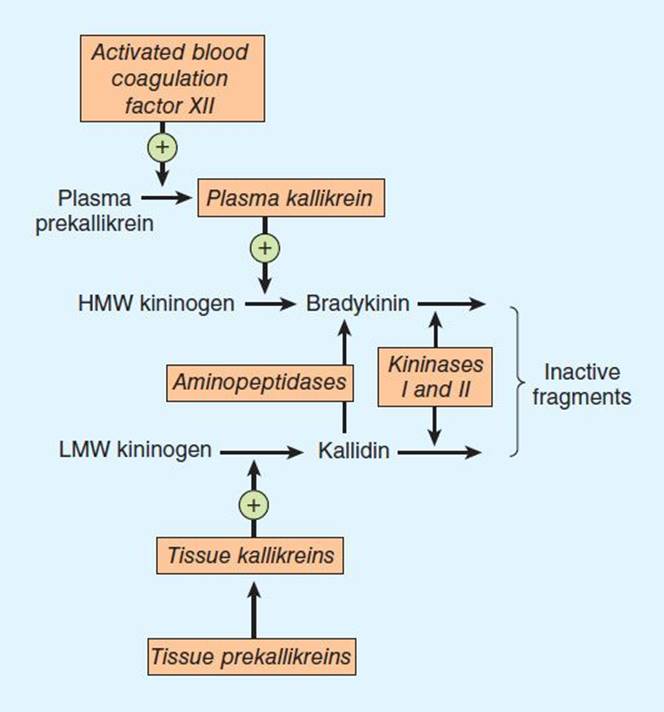
FIGURE 17–4 The kallikrein-kinin system. Kininase II is identical to converting enzyme peptidyl dipeptidase (ACE).
Kinins are metabolized rapidly (half-life < 15 seconds) by nonspecific exopeptidases or endopeptidases, commonly referred to as kininases. Two plasma kininases have been characterized. Kininase I, apparently synthesized in the liver, is a carboxypeptidase that releases the carboxyl terminal arginine residue. Kininase II is present in plasma and vascular endothelial cells throughout the body. It is identical to angiotensin-converting enzyme (ACE, peptidyl dipeptidase), discussed above. Kininase II inactivates kinins by cleaving the carboxyl terminal dipeptide phenylalanyl-arginine. Like angiotensin I, bradykinin is almost completely hydrolyzed during a single passage through the pulmonary vascular bed.
PHYSIOLOGIC & PATHOLOGIC EFFECTS OF KININS
Effects on the Cardiovascular System
Kinins produce marked arteriolar dilation in several vascular beds, including the heart, skeletal muscle, kidney, liver, and intestine. In this respect, kinins are approximately 10 times more potent on a molar basis than histamine. The vasodilation may result from a direct inhibitory effect of kinins on arteriolar smooth muscle or may be mediated by the release of nitric oxide or vasodilator prostaglandins such as PGE2 and PGI2. In contrast, the predominant effect of kinins on veins is contraction; again, this may result from direct stimulation of venous smooth muscle or from the release of venoconstrictor prostaglandins such as PGF2α. Kinins also produce contraction of most visceral smooth muscle.
When injected intravenously, kinins produce a rapid but brief fall in blood pressure that is due to their arteriolar vasodilator action. Intravenous infusions of the peptide fail to produce a sustained decrease in blood pressure; prolonged hypotension can only be produced by progressively increasing the rate of infusion. The rapid reversibility of the hypotensive response to kinins is due primarily to reflex increases in heart rate, myocardial contractility, and cardiac output. In some species, bradykinin produces a biphasic change in blood pressure—an initial hypotensive response followed by an increase above the preinjection level. The increase in blood pressure may be due to a reflex activation of the sympathetic nervous system, but under some conditions, bradykinin can directly release catecholamines from the adrenal medulla and stimulate sympathetic ganglia. Bradykinin also increases blood pressure when injected into the central nervous system, but the physiologic significance of this effect is not clear, since it is unlikely that kinins cross the blood-brain barrier. (Note, however, that bradykinin can increase the permeability of the blood-brain barrier to some other substances.) Kinins have no consistent effect on sympathetic or parasympathetic nerve endings.
The arteriolar dilation produced by kinins causes an increase in pressure and flow in the capillary bed, thus favoring efflux of fluid from blood to tissues. This effect may be facilitated by increased capillary permeability resulting from contraction of endothelial cells and widening of intercellular junctions, and by increased venous pressure secondary to constriction of veins. As a result of these changes, water and solutes pass from the blood to the extracellular fluid, lymph flow increases, and edema may result.
The role that endogenous kinins play in the regulation of blood pressure is not clear. They do not appear to participate in the control of blood pressure under resting conditions but may play a role in postexercise hypotension.
Effects on Endocrine & Exocrine Glands
As noted earlier, prekallikreins and kallikreins are present in several glands, including the pancreas, kidney, intestine, salivary glands, and sweat glands, and they can be released into the secretory fluids of these glands. The function of the enzymes in these tissues is not known. Since kinins have such marked effects on smooth muscle, they may modulate the tone of salivary and pancreatic ducts, help regulate gastrointestinal motility, and act as local modulators of blood flow. Kinins also influence the transepithelial transport of water, electrolytes, glucose, and amino acids, and may regulate the transport of these substances in the gastrointestinal tract and kidney. Finally, kallikreins may play a role in the physiologic activation of certain prohormones, including proinsulin and prorenin.
Role in Inflammation & Pain
Bradykinin has long been known to produce the four classic symptoms of inflammation—redness, local heat, swelling, and pain. Kinins are rapidly generated after tissue injury and play a pivotal role in the development and maintenance of these inflammatory processes.
Kinins are potent pain-producing substances when applied to a blister base or injected intradermally. They elicit pain by stimulating nociceptive afferents in the skin and viscera.
Role in Hereditary Angioedema
Hereditary angioedema is a rare autosomal dominant disorder that results from deficiency or dysfunction of the C1 esterase inhibitor (C1-INH), a major inhibitor of proteases of the complement, coagulation, and kallikrein-kinin systems. C1-INH deficiency results in activation of kallikrein and increased formation of bradykinin, which by increasing vascular permeability and other actions, causes recurrent episodes of angioedema of the airways, gastrointestinal tract, extremities, and genitalia. Hereditary angioedema can be treated with drugs that inhibit the formation or actions of bradykinin (see below).
Other Effects
There is evidence that bradykinin may play a beneficial, protective role in certain cardiovascular diseases and ischemic stroke-induced brain injury. On the other hand, it has been implicated in cancer and some central nervous system diseases.
KININ RECEPTORS & MECHANISMS OF ACTION
The biologic actions of kinins are mediated by specific receptors located on the membranes of the target tissues. Two types of kinin receptors, termed B1 and B2, have been defined based on the rank orders of agonist potencies; both are G protein-coupled receptors. (Note that B here stands for bradykinin, not for β adrenoceptor.) Bradykinin displays the highest affinity in most B2 receptor systems, followed by Lys-bradykinin. One exception is the B2 receptor that mediates contraction of venous smooth muscle; this appears to be more sensitive to Lys-bradykinin. Recent evidence suggests the existence of two B2-receptor subtypes, which have been termed B2A and B2B.
B1 receptors appear to have a very limited distribution in mammalian tissues and have few known functional roles. Studies with knockout mice that lack functional B1 receptors suggest that these receptors participate in the inflammatory response and may also be important in long-lasting kinin effects such as collagen synthesis and cell multiplication. By contrast, B2 receptors have a widespread distribution that is consistent with the multitude of biologic effects that are mediated by this receptor type. Agonist binding to B2 receptors sets in motion multiple signal transduction events, including calcium mobilization, chloride transport, formation of nitric oxide, and activation of phospholipase C, phospholipase A2, and adenylyl cyclase.
DRUGS AFFECTING THE KALLIKREIN-KININ SYSTEM
Drugs that modify the activity of the kallikrein-kinin system are available. Considerable effort has been directed toward developing kinin receptor antagonists, since such drugs have considerable therapeutic potential as anti-inflammatory and antinociceptive agents. Competitive antagonists of both B1 and B2 receptors are available for research use. Examples of B1 receptor antagonists are the peptides [Leu8-des-Arg9]bradykinin and Lys[Leu8-des-Arg9]bradykinin. The first B2 receptor antagonists to be discovered were also peptide derivatives of bradykinin. These first-generation antagonists were used extensively in animal studies of kinin receptor pharmacology. However, their half-life is short, and they are almost inactive on the human B2 receptor.
Icatibant is a second-generation B2 receptor antagonist. It is a decapeptide with an affinity for the B2 receptor similar to that of bradykinin and is absorbed rapidly after subcutaneous administration. Icatibant has been shown to be effective in the treatment of hereditary angioedema. It may also be useful in other conditions including drug-induced angioedema, airway disease, thermal injury, ascites, and pancreatitis.
Recently, a third generation of B2-receptor antagonists was developed; examples are FR 173657, FR 172357, and NPC 18884. These antagonists block both human and animal B2 receptors and are orally active. They have been reported to inhibit bradykinin-induced bronchoconstriction in guinea pigs, carrageenin-induced inflammation in rats, and capsaicin-induced nociception in mice. These antagonists have promise for the treatment of inflammatory pain in humans.
SSR240612 is a new, potent, and orally active selective antagonist of B1 receptors in humans and several animal species. It exhibits analgesic and anti-inflammatory activities in mice and rats and is currently in preclinical development for the treatment of inflammatory and neurogenic pain.
The synthesis of kinins can be inhibited with the kallikrein inhibitor aprotinin. Kinin synthesis can also be inhibited by two preparations of human plasma C1-INH, cinryze and berinert, and these are used for the intravenous prophylaxis or treatment of hereditary angioedema. Ecallantide, a more recently developed recombinant plasma kallikrein inhibitor, is also effective. It is more potent and selective than C1-INH and can be administered by subcutaneous injection.
Actions of kinins mediated by prostaglandin generation can be blocked nonspecifically with inhibitors of prostaglandin synthesis such as aspirin. Conversely, the actions of kinins can be enhanced with ACE inhibitors, which block the degradation of the peptides. Indeed, as noted above, inhibition of bradykinin metabolism by ACE inhibitors contributes significantly to their antihypertensive action.
Selective B2 agonists are under study and have been shown to be effective in some animal models of human cardiovascular disease. These drugs have potential for the treatment of hypertension, myocardial hypertrophy, and other diseases.
![]() VASOPRESSIN
VASOPRESSIN
Vasopressin (arginine vasopressin, AVP; antidiuretic hormone, ADH) plays an important role in the long-term control of blood pressure through its action on the kidney to increase water reabsorption. This and other aspects of the physiology of AVP are discussed in Chapters 15 and 37 and will not be reviewed here.
AVP also plays an important role in the regulation of arterial pressure by its vasoconstrictor action. Mutant mice lacking the V1a receptor gene show significantly lower blood pressure compared with control mice. AVP increases total peripheral resistance when infused in doses less than those required to produce maximum urine concentration. Such doses do not normally increase arterial pressure because the vasopressor activity of the peptide is buffered by a reflex decrease in cardiac output. When the influence of this reflex is removed, eg, in shock, pressor sensitivity to AVP is greatly increased. Pressor sensitivity to AVP is also enhanced in patients with idiopathic orthostatic hypotension. Higher doses of AVP increase blood pressure even when baroreceptor reflexes are intact.
VASOPRESSIN RECEPTORS, AGONISTS, & ANTAGONISTS
Three subtypes of AVP receptors have been identified; all are G protein-coupled. V1a receptors mediate the vasoconstrictor action of AVP; V1b receptors mediate release of ACTH by pituitary corticotropes; and V2 receptors mediate the antidiuretic action. V1a effects are mediated by Gq activation of phospholipase C, formation of inositol trisphosphate, and increased intracellular calcium concentration. V2 effects are mediated by Gs activation of adenylyl cyclase.
AVP analogs selective for vasoconstrictor or antidiuretic activity have been synthesized. The most specific V1 vasoconstrictor agonist synthesized to date is [Phe2, Ile3, Orn8]vasotocin. Selective V2antidiuretic analogs include 1-deamino[D-Arg8]arginine vasopressin (dDAVP) and 1-deamino[Val4,D-Arg8]arginine vasopressin (dVDAVP).
AVP has proved beneficial in the treatment of vasodilatory shock states, at least in part by virtue of its V1a agonist activity. Terlipressin (triglycyl lysine vasopressin), a synthetic vasopressin analog that is converted to lysine vasopressin in the body, is also effective. Terlipressin and [Phe2, Ile3, Orn8]vasotocin may have advantages over AVP because they are more selective for V1 receptors and have longer half-lives.
Antagonists of the vasoconstrictor action of AVP are also available. The peptide antagonist d(CH2)5[Tyr(Me)2]AVP also has antioxytocic activity but does not antagonize the antidiuretic action of AVP. A related antagonist d(CH2)5[Tyr(Me)2 Dab5]AVP lacks oxytocin antagonism but has less anti-V1 activity. Nonpeptide, orally active V1a-receptor antagonists have been developed, examples being relcovaptanand SRX251.
The V1a antagonists have been particularly useful in revealing the important role that AVP plays in blood pressure regulation in situations such as dehydration and hemorrhage. They have potential as therapeutic agents for the treatment of such diverse conditions as Raynaud’s disease, hypertension, heart failure, brain edema, motion sickness, cancer, preterm labor, and anger reduction. To date, most studies have focused on heart failure; promising results have been obtained with V2 antagonists such as tolvaptan, which is, however, currently approved only for use in hyponatremia. V1a antagonists also have potential, and conivaptan (YM087), a drug with both V1a and V2 antagonist activity, has also been approved for treatment of hyponatremia (see Chapter 15).
![]() NATRIURETIC PEPTIDES
NATRIURETIC PEPTIDES
Synthesis & Structure
The atria and other tissues of mammals contain a family of peptides with natriuretic, diuretic, vasorelaxant, and other properties. The family includes atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP). The peptides share a common 17-amino-acid disulfide ring with variable C- and N-terminals (Figure 17–5). A fourth peptide, urodilatin, has the same structure as ANP with an extension of four amino acids at the N-terminal. The renal effects of these peptides are discussed in Chapter 15.
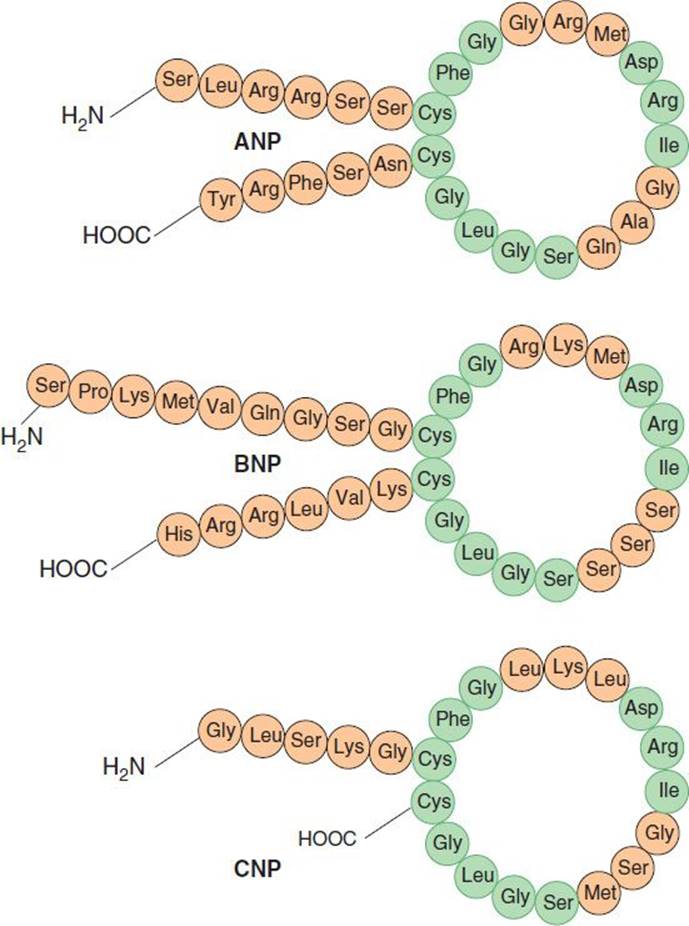
FIGURE 17–5 Structures of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP). Sequences common to the peptides are indicated in green.
ANP is derived from the carboxyl terminal end of a common precursor termed preproANP. ANP is synthesized primarily in cardiac atrial cells, but it is also synthesized in ventricular myocardium, by neurons in the central and peripheral nervous systems, and in the lungs.
The most important stimulus to the release of ANP from the heart is atrial stretch via mechanosensitive ion channels. ANP release is also increased by volume expansion, changing from the standing to the supine position, and exercise. ANP release can also be increased by sympathetic stimulation via α1A adrenoceptors, endothelins via the ETA-receptor subtype (see below), glucocorticoids, and AVP. Plasma ANP concentration increases in several pathologic states, including heart failure, primary aldosteronism, chronic renal failure, and inappropriate ADH secretion syndrome.
Administration of ANP increases sodium excretion and urine flow. The ANP-induced natriuresis is due both to an increase in glomerular filtration rate and a decrease in proximal tubular sodium reabsorption. ANP also inhibits the release of renin, aldosterone, and AVP; these changes may also increase sodium and water excretion. Finally, ANP causes vasodilation and decreases arterial blood pressure. Suppression of ANP production or blockade of its action impairs the natriuretic response to volume expansion, and increases blood pressure.
Like ANP, BNP is synthesized primarily in the heart. It exists in two forms, with either 26 or 32 amino acids (Figure 17–5). Like ANP, the release of BNP appears to be volume related; indeed, the two peptides may be co-secreted. BNP exhibits natriuretic, diuretic, and hypotensive activities similar to those of ANP but circulates at a lower concentration.
CNP consists of 22 amino acids (Figure 17–5). It is located predominantly in the central nervous system but is also present in other tissues including the vascular endothelium, kidneys, and intestine. It has not been found in significant concentrations in the circulation. CNP has less natriuretic and diuretic activity than ANP and BNP but is a potent vasodilator and may play a role in the regulation of peripheral resistance.
Urodilatin is synthesized in the distal tubules of the kidneys by alternative processing of the ANP precursor. It elicits potent natriuresis and diuresis, and thus functions as a paracrine regulator of sodium and water excretion. It also relaxes vascular smooth muscle.
Pharmacodynamics & Pharmacokinetics
The biologic actions of the natriuretic peptides are mediated through association with specific high-affinity receptors located on the surface of the target cells. Three receptor subtypes termed ANPA, ANPB,and ANPC (also known as NPR1, NPR2, and NPR3) have been identified. The ANPA and ANPB receptors contain guanylyl cyclase activity at their intracellular domains. The primary ligands of the ANPAreceptor are ANP and BNP. The ANPB receptor is similar in structure to the ANPA receptor, but its primary ligand appears to be CNP. The ANPC receptor is coupled to inhibition of adenylyl cyclase or activation of phospholipase C; it binds all three natriuretic peptides.
The natriuretic peptides have a short half-life in the circulation. They are metabolized in the kidneys, liver, and lungs by the neutral endopeptidase NEP 24.11. Inhibition of this endopeptidase results in increases in circulating levels of the natriuretic peptides, natriuresis, and diuresis. The peptides are also removed from the circulation by binding to ANPC receptors in the vascular endothelium. This receptor binds the natriuretic peptides with equal affinity. The receptor and bound peptide are internalized, the peptide is degraded enzymatically, and the receptor is returned to the cell surface. Patients with heart failure have high plasma levels of ANP and BNP; the latter has emerged as a diagnostic and prognostic marker in this condition.
CLINICAL ROLE OF NATRIURETIC PEPTIDES
Natriuretic peptides may be administered as recombinant ANP (carperitide) or BNP (nesiritide). These peptides produce vasodilation, natriuresis, and inhibition of the renin-angiotensin system. These actions appear very promising for the treatment of congestive heart failure, but clinical studies have produced variable results. The serum concentration of endogenous BNP rises in heart failure, and monitoring this peptide has been shown to have prognostic value.
The circulating levels of natriuretic peptides can also be increased by vasopeptidase inhibitors. These constitute a novel class of cardiovascular drugs that inhibit two metalloprotease enzymes, NEP 24.11 and ACE. They thus simultaneously increase the levels of natriuretic peptides and decrease the formation of ANG II. As a result, they enhance vasodilation, reduce vasoconstriction, and increase sodium excretion, in turn reducing peripheral vascular resistance and blood pressure.
Recently developed vasopeptidase inhibitors include omapatrilat, sampatrilat, and fasidotrilat. Omapatrilat, which has received the most attention, lowers blood pressure in animal models of hypertension as well as in hypertensive patients, and improves cardiac function in patients with heart failure. Unfortunately, omapatrilat causes a significant incidence of angioedema in addition to cough and dizziness and has not been approved for clinical use.
![]() ENDOTHELINS
ENDOTHELINS
The endothelium is the source of a variety of substances with vasodilator (PGI2 and nitric oxide) and vasoconstrictor activities. The latter include the endothelin family, potent vasoconstrictor peptides that were first isolated from aortic endothelial cells.
Biosynthesis, Structure, & Clearance
Three isoforms of endothelin have been identified: the originally described endothelin, ET-1, and two similar peptides, ET-2 and ET-3. Each isoform is a product of a different gene and is synthesized as a prepro form that is processed to a propeptide and then to the mature peptide. Processing to the mature peptides occurs through the action of endothelin-converting enzyme. Each endothelin is a 21-amino-acid peptide containing two disulfide bridges. The structure of ET-1 is shown in Figure 17–6.
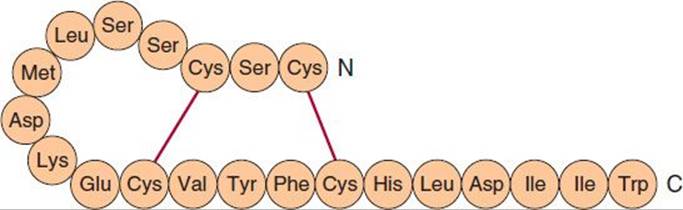
FIGURE 17–6 Structure of human endothelin-1.
Endothelins are widely distributed in the body. ET-1 is the predominant endothelin secreted by the vascular endothelium. It is also produced by neurons and astrocytes in the central nervous system and in endometrial, renal mesangial, Sertoli, breast epithelial, and other cells. ET-2 is produced predominantly in the kidneys and intestine, whereas ET-3 is found in highest concentration in the brain but is also present in the gastrointestinal tract, lungs, and kidneys. Endothelins are present in the blood but in low concentration; they apparently act locally in a paracrine or autocrine fashion rather than as circulating hormones.
The expression of the ET-1 gene is increased by growth factors and cytokines, including transforming growth factor-β (TGF-β) and interleukin 1 (IL-1), vasoactive substances including ANG II and AVP, and mechanical stress. Expression is inhibited by nitric oxide, prostacyclin, and ANP.
Clearance of endothelins from the circulation is rapid and involves both enzymatic degradation by NEP 24.11 and clearance by the ETB receptor.
Actions
Endothelins exert widespread actions in the body. In particular, they cause potent dose-dependent vasoconstriction in most vascular beds. Intravenous administration of ET-1 causes a rapid and transient decrease in arterial blood pressure followed by a sustained increase. The depressor response results from release of prostacyclin and nitric oxide from the vascular endothelium, whereas the pressor response is due to direct contraction of vascular smooth muscle. Endothelins also exert direct positive inotropic and chronotropic actions on the heart and are potent coronary vasoconstrictors. They act on the kidneys to cause vasoconstriction and decrease glomerular filtration rate and sodium and water excretion. In the respiratory system, they cause potent contraction of tracheal and bronchial smooth muscle. Endothelins interact with several endocrine systems, increasing the secretion of renin, aldosterone, AVP, and ANP. They exert a variety of actions on the central and peripheral nervous systems, the gastrointestinal system, the liver, the urinary tract, the reproductive system, eye, skeletal system, and skin. ET-1 is a potent mitogen for vascular smooth muscle cells, cardiac myocytes, and glomerular mesangial cells.
Two endothelin receptor subtypes, termed ETA and ETB, are widespread in the body. ETA receptors have a high affinity for ET-1 and a low affinity for ET-3 and are located on smooth muscle cells, where they mediate vasoconstriction (Figure 17–7). ETB receptors have approximately equal affinities for ET-1 and ET-3 and are primarily located on vascular endothelial cells, where they mediate release of PGI2 and nitric oxide. Some ETB receptors are also present on smooth muscle cells and mediate vasoconstriction. Both receptor subtypes belong to the G protein-coupled seven-transmembrane domain family of receptors.
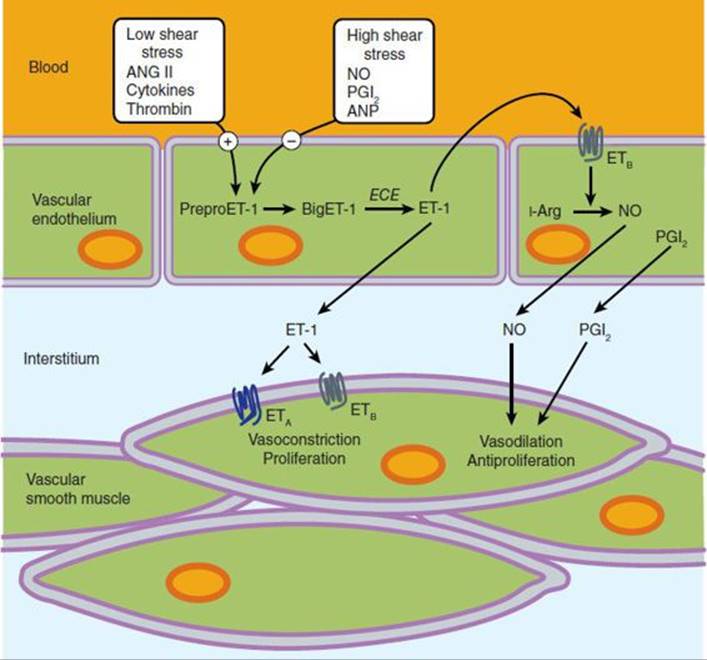
FIGURE 17–7 Generation of endothelin-1 (ET-1) in the vascular endothelium, and its direct and indirect effects on smooth muscle cells mediated by ETA and ETB receptors. ANG II, angiotensin II; ANP, atrial natriuretic peptide; Arg, arginine; BigET-1, proET-1; ECE, endothelial- converting enzyme; NO, nitric oxide; PreproET-1, precursor of BigET-1; PGI2, prostaglandin I2.
The signal transduction mechanisms triggered by binding of ET-1 to its vascular receptors include stimulation of phospholipase C, formation of inositol trisphosphate, and release of calcium from the endoplasmic reticulum, which results in vasoconstriction. Conversely, stimulation of PGI2 and nitric oxide synthesis results in decreased intracellular calcium concentration and vasodilation.
INHIBITORS OF ENDOTHELIN SYNTHESIS & ACTION
The endothelin system can be blocked with receptor antagonists and drugs that block endothelin-converting enzyme. Endothelin ETA or ETB receptors can be blocked selectively, or both can be blocked with nonselective ETA-ETBantagonists.
Bosentan is a nonselective receptor blocker. It is active orally, and blocks both the initial transient depressor (ETB) and the prolonged pressor (ETA) responses to intravenous endothelin. Many orally active endothelin receptor antagonists with increased selectivity have been developed and are available for research use. Examples include the selective ETA antagonists ambrisentan, which has been approved by the Food and Drug Administration to treat pulmonary artery hypertension, and sitaxsentan. A new dual endothelin receptor antagonist, macitentan, has recently been approved by the Food and Drug Administration. It appears to have increased efficacy in pulmonary hypertension compared with the other antagonists and is well tolerated with fewer side effects.
The formation of endothelins can be blocked by inhibiting endothelin-converting enzyme with phosphoramidon. Phosphoramidon is not specific for endothelin-converting enzyme, but more selective inhibitors including CGS35066 are now available for research. Although the therapeutic potential of these drugs appeared similar to that of the endothelin receptor antagonists (see below), their use has been eclipsed by endothelin antagonists.
Physiologic & Pathologic Roles of Endothelin: Effects of Endothelin Antagonists
Systemic administration of endothelin receptor antagonists or endothelin-converting enzyme inhibitors causes vasodilation and decreases arterial pressure in humans and experimental animals. Intra-arterial administration of the drugs also causes slow-onset forearm vasodilation in humans. These observations provide evidence that the endothelin system participates in the regulation of vascular tone, even under resting conditions. The activity of the system is higher in males than in females. It increases with age, an effect that can be counteracted by regular aerobic exercise.
Increased production of ET-1 has been implicated in a variety of cardiovascular diseases, including hypertension, cardiac hypertrophy, heart failure, atherosclerosis, coronary artery disease, and myocardial infarction. ET-1 also participates in pulmonary diseases, including asthma and pulmonary hypertension; renal diseases; and several malignancies, including ovarian cancer.
Endothelin antagonists have considerable potential for the treatment of these diseases. Indeed, endothelin antagonism with bosentan, sitaxsentan, ambrisentan, and macitentan has proved to be a moderately effective and generally well-tolerated treatment for patients with pulmonary arterial hypertension, an important condition with few effective treatments. Unfortunately, both bosentan and sitaxsentan have been associated with severe hepatic toxicity. Other promising targets for these drugs are resistant hypertension, chronic renal disease, connective tissue disease, and subarachnoid hemorrhage. On the other hand, clinical trials of the drugs in the treatment of congestive heart failure have been disappointing.
Endothelin antagonists occasionally cause systemic hypotension, increased heart rate, facial flushing or edema, and headaches. Potential gastrointestinal effects include nausea, vomiting, and constipation. Because of their teratogenic effects, endothelin antagonists are contraindicated in pregnancy. Bosentan has been associated with fatal hepatotoxicity, and patients taking this drug must have monthly liver function tests. Negative pregnancy test results are required for women of child-bearing age to take this drug.
![]() VASOACTIVE INTESTINAL PEPTIDE
VASOACTIVE INTESTINAL PEPTIDE
Vasoactive intestinal peptide (VIP) is a 28-amino-acid peptide that belongs to the glucagon-secretin family of peptides. VIP is widely distributed in the central and peripheral nervous systems, where it functions as one of the major peptide neurotransmitters. It is present in cholinergic presynaptic neurons in the central nervous system, and in peripheral peptidergic neurons innervating diverse tissues including the heart, lungs, gastrointestinal and urogenital tracts, skin, eyes, ovaries, and thyroid gland. Many blood vessels are innervated by VIP neurons. VIP is also present in key organs of the immune system including the thymus, spleen, and lymph nodes. Although VIP is present in blood, where it undergoes rapid degradation, it does not appear to function as a hormone. VIP participates in a wide variety of biologic functions including metabolic processes, secretion of endocrine and exocrine glands, cell differentiation, smooth muscle relaxation, and modulation of the immune response.
VIP exerts significant effects on the cardiovascular system. It produces marked vasodilation in most vascular beds and in this regard is more potent on a molar basis than acetylcholine. In the heart, VIP causes coronary vasodilation and exerts positive inotropic and chronotropic effects. It may thus participate in the regulation of coronary blood flow, cardiac contraction, and heart rate.
The Treatment of Pulmonary Hypertension
Idiopathic pulmonary arterial hypertension (PAH) is a progressive and potentially fatal condition; signs and symptoms include dyspnea, chest pain, syncope, cardiac arrhythmias, and right heart failure. Continuous nasal oxygen supplementation is required for most patients and anticoagulants are commonly used. Medical treatments directed at elevated pulmonary vascular resistance have been less successful than those used in ordinary hypertension (see Chapter 11). In addition to the endothelin antagonists mentioned in the text (bosentan, ambrisentan, and macitentan are approved for use in PAH), vasoactive agents that have been promoted for PAH include prostaglandins (epoprostenol, treprostinil, iloprost), nitric oxide, PDE-5 inhibitors (sildenafil, tadalafil), and Ca2+ channel blockers (nifedipine, amlodipine, diltiazem). Riociguat, a small molecule activator of soluble guanylyl cyclase, increases cGMP independently of nitric oxide, reduces pulmonary vascular pressure, and increases exercise duration. Riociguat was approved in the USA in 2013. Fasudil is an investigational selective RhoA/Rho kinase (ROCK) inhibitor that appears to reduce pulmonary artery pressure in PAH. Surgical treatment for advanced disease includes creation of a right atrial to left atrial shunt and lung transplantation.
The effects of VIP are mediated by two G protein-coupled receptors, VPAC1 and VPAC2. Both receptors are widely distributed in the central nervous system and in the heart, blood vessels, and other tissues. VIP has a high affinity for both receptor subtypes. Binding of VIP to its receptors results in activation of adenylyl cyclase and formation of cAMP, which is responsible for the vasodilation and many other effects of the peptide. Other actions may be mediated by inositol trisphosphate synthesis and calcium mobilization. VIP can also bind with low affinity to the VIP-like peptide pituitary adenylyl cyclase-activating peptide receptor, PAC1.
VIP analogs with longer half-lives than VIP are now available for research use. An example is stearyl-Nle17-VIP, which is 100 times more potent than the native peptide. These drugs have potential as therapeutic agents for cardiovascular, pulmonary, gastrointestinal, and nervous system diseases including Alzheimer’s and Parkinson’s disease. They may also be effective in treating various inflammatory diseases and diabetes. Indeed, some VIP derivatives are currently undergoing preclinical and clinical testing for the treatment of type 2 diabetes and chronic obstructive pulmonary disease. Unfortunately, their use is currently limited by several issues including poor oral availability and hypotension. VIP receptor antagonists have also been developed.
![]() SUBSTANCE P
SUBSTANCE P
Substance P belongs to the tachykinin family of peptides, which share the common carboxyl terminal sequence Phe-Gly-Leu-Met. Other members of this family are neurokinin A and neurokinin B.Substance P is an undecapeptide, while neurokinins A and B are decapeptides.
Substance P is present in the central nervous system, where it is a neurotransmitter (see Chapter 21), and in the gastrointestinal tract, where it may play a role as a transmitter in the enteric nervous system and as a local hormone (see Chapter 6).
Substance P is the most important member of the tachykinin family. It exerts a variety of central actions that implicate the peptide in behavior, anxiety, depression, nausea, and emesis; it is present in peripheral afferent pain fibers. It is a potent arteriolar vasodilator, producing marked hypotension in humans and several animal species. The vasodilation is mediated by release of nitric oxide from the endothelium. Substance P causes contraction of venous, intestinal, and bronchial smooth muscle. It stimulates secretion by the salivary glands and causes diuresis and natriuresis by the kidneys.
The actions of substance P and neurokinins A and B are mediated by three Gq protein-coupled tachykinin receptors designated NK1, NK2, and NK3. Substance P is the preferred ligand for the NK1 receptor. This receptor is widespread throughout the body and is the predominant tachykinin receptor in the human brain. However, neurokinins A and B also possess considerable affinity for this receptor. In humans, most of the central and peripheral effects of substance P are mediated by NK1 receptors. All three receptor subtypes are coupled to inositol trisphosphate synthesis and calcium mobilization.
Several nonpeptide NK1 receptor antagonists have been developed. These compounds are highly selective and orally active, and enter the brain. Recent clinical trials have shown that these antagonists may be useful in treating depression and other disorders and in preventing chemotherapy-induced emesis. The first of these to be approved for the prevention of chemotherapy-induced and postoperative nausea and vomiting is aprepitant (see Chapter 62). Fosaprepitant is a prodrug that is converted to aprepitant after intravenous administration and may be a useful parenteral alternative to oral aprepitant.
Recent studies have implicated the substance P-NK1 system in cancer. Substance P and NK1 receptors are present in a variety of tumor cells, and NK1 receptor antagonists exert an antitumor action. Thus, drugs such as aprepitant have potential as anti-cancer agents.
![]() NEUROTENSIN
NEUROTENSIN
Neurotensin (NT) is a tridecapeptide that was first isolated from the central nervous system but subsequently was found to be present in the gastrointestinal tract. It is also present in the circulation and in several organs including the heart, lungs, liver, pancreas, and spleen.
NT is synthesized as part of a larger precursor that also contains neuromedin N, a six-amino-acid NT-like peptide. In the brain, processing of the precursor leads primarily to the formation of NT and neuromedin N; these are released together from nerve endings. In the gut, processing leads mainly to the formation of NT and a larger peptide that contains the neuromedin N sequence at the carboxyl terminal. Both peptides are secreted into the circulation after ingestion of food. Most of the activity of NT is mediated by the last six amino acids, NT(8-13).
Like many other neuropeptides, NT serves a dual function as a neurotransmitter or neuromodulator in the central nervous system and as a local hormone in the periphery. When administered centrally, NT exerts potent effects including hypothermia, antinociception, and modulation of dopamine and glutamate neurotransmission. When administered into the peripheral circulation, it causes vasodilation, hypotension, increased vascular permeability, increased secretion of several anterior pituitary hormones, hyperglycemia, inhibition of gastric acid and pepsin secretion, and inhibition of gastric motility. It also exerts effects on the immune system.
In the central nervous system, there are close associations between NT and dopamine systems, and NT may be involved in clinical disorders involving dopamine pathways such as schizophrenia, Parkinson’s disease, and drug abuse. Consistent with this, it has been shown that central administration of NT produces effects in rodents similar to those produced by antipsychotic drugs.
The effects of NT are mediated by three subtypes of NT receptors, designated NTR1, NTR2, and NTR3, also known as NTS1, NTS2, and NTS3. NTR1 and NTR2 receptors belong to the Gq protein-coupled superfamily; the NTR3receptor is a single transmembrane protein that is structurally unrelated to NTR1 or NTR2. It belongs to a family of sorting proteins and is therefore known as NTR3/sortilin.
The potential use of NT as an antipsychotic agent has been hampered by its rapid degradation in the circulation and inability to cross the blood-brain barrier. However, a series of analogs of NT(8-13) that exert antipsychotic-like activity in animal studies has been developed. These agonists include NT69L, which binds with high affinity to NTR1 and NTR2; and NT79, which preferentially binds to NTR2. Another agonist, PD149163, has improved metabolic stability.
In addition to their possible role as antipsychotic drugs, these agonists may be useful in the treatment of pain, psychostimulant abuse, and Parkinson’s disease. Potential adverse effects include hypothermia and hypotension. Development of tolerance to some of the effects of the agonists may occur.
NT receptors can be blocked with the nonpeptide antagonists SR142948A and meclinertant (SR48692). SR142948A is a potent antagonist of the hypothermia and analgesia produced by centrally administered NT. It also blocks the cardiovascular effects of systemic NT.
![]() CALCITONIN GENE-RELATED PEPTIDE
CALCITONIN GENE-RELATED PEPTIDE
Calcitonin gene-related peptide (CGRP) is a member of the calcitonin family of peptides, which also includes calcitonin, adrenomedullin, and amylin. CGRP consists of 37 amino acids. In humans, CGRP exists in two forms termed α-CGRP and β-CGRP, which are derived from separate genes and differ by three amino acids but exhibit similar biological activity. Like calcitonin, CGRP is present in large quantities in the C cells of the thyroid gland. It is also distributed widely in the central and peripheral nervous systems, cardiovascular and respiratory systems, and gastrointestinal tract. In the cardiovascular system, CGRP-containing neuronal fibers are more abundant around arteries than around veins and in atria than in ventricles. CGRP fibers are associated with most smooth muscles of the gastrointestinal tract. CGRP is found with substance P (see above) in some of these regions and with acetylcholine in others.
When CGRP is injected into the central nervous system, it produces a variety of effects, including hypertension and suppression of feeding. When injected into the systemic circulation, the peptide causes hypotension and tachycardia. The hypotensive action of CGRP results from the potent vasodilator action of the peptide; indeed, CGRP is the most potent vasodilator yet discovered. It dilates multiple vascular beds, but the coronary circulation is particularly sensitive. The vasodilation is mediated via a nonendothelial mechanism through activation of adenylyl cyclase.
The actions of CGRP are mediated via a single receptor type. This heterodimeric receptor consists of the G protein-coupled calcitonin receptor-like receptor (CLR) combined with the receptor activity-modifying protein RAMP1.
Peptide and nonpeptide antagonists of the CGRP receptor have been developed. CGRP8-37 has been used extensively to investigate the actions of CGRP but displays affinity for other related receptors including those for adrenomedullin (see below). Nonpeptide CGRP receptor antagonists target the interface between CLR and RAMP1 and thereby make them more selective for the CGRP receptor. Examples are olcegepant and telcagepant.
Evidence is accumulating that release of CGRP from trigeminal nerves plays a central role in the pathophysiology of migraine. The peptide is released during migraine attacks, and successful treatment of migraine with a selective serotonin agonist normalizes cranial CGRP levels. Clinical trials showed olcegepant to be effective in treating migraine, but because of its low bioavailability, it has to be administered by intravenous injection. Telcagepant is also effective and is orally active but has exhibited liver toxicity in a small number of patients.
![]() ADRENOMEDULLIN
ADRENOMEDULLIN
Adrenomedullin (AM) was first discovered in human adrenal medullary pheochromocytoma tissue. It is a 52-amino-acid peptide with a six-amino-acid ring and a C-terminal amidation sequence. Like CGRP, AM is a member of the calcitonin family of peptides. A related peptide termed adrenomedullin 2, also called intermedin, has been identified in humans and other mammals.
AM is widely distributed in the body. The highest concentrations are found in the adrenal glands, hypothalamus, and anterior pituitary, but high levels are also present in the kidneys, lungs, cardiovascular system, and gastrointestinal tract. AM in plasma apparently originates in the heart and vasculature.
In animals, AM dilates resistance vessels in the kidney, brain, lung, hind limbs, and mesentery, resulting in a marked, long-lasting hypotension. The hypotension in turn causes reflex increases in heart rate and cardiac output. These responses also occur during intravenous infusion of the peptide in healthy human subjects. AM also acts on the kidneys to increase sodium excretion and renin release, and it exerts other endocrine effects including inhibition of aldosterone and insulin secretion. It acts on the central nervous system to increase sympathetic outflow.
The diverse actions of AM are mediated by a receptor closely related to the CGRP receptor (see above). CLR co-assembles with RAMP subtypes 2 and 3, thus forming the AM receptor. Binding of AM to CLR activates Gs and triggers cAMP formation in vascular smooth muscle cells, and increases nitric oxide production in endothelial cells. Other signaling pathways are also involved.
Circulating AM levels increase during intense exercise. They also increase in a number of pathologic states, including essential and pulmonary hypertension, acute myocardial infarction, and cardiac and renal failure. Plasma AM levels are increased in proportion to the severity of these diseases and this can be a useful prognostic marker. The roles of AM in these states remain to be defined, but it is currently thought that the peptide functions as a physiologic antagonist of the actions of vasoconstrictors including ET-1 and ANG II. By virtue of these actions, AM may protect against cardiovascular overload and injury, and AM may be beneficial in the treatment of some cardiovascular diseases.
![]() NEUROPEPTIDE Y
NEUROPEPTIDE Y
The neuropeptide Y family is a multiligand/multireceptor system consisting of three polypeptide agonists that bind and activate four distinct receptors with different affinity and potency. The peptides are pancreatic polypeptide (PP), peptide YY (PYY), and neuropeptide Y (NPY). Each peptide consists of 36 amino acids and has an amidated C-terminus. PP is secreted by the islets of Langerhans after food ingestion in proportion to the caloric content and appears to act mainly in the brainstem and vagus to promote appetite suppression, inhibit gastric emptying, and increase energy expenditure; it also exerts direct actions in the gut. PYY is released by entero-endocrine L cells of the distal gut in proportion to food intake and produces anorexigenic effects.
NPY is one of the most abundant neuropeptides in both the central and peripheral nervous systems. Whereas PYY and PP act as neuroendocrine hormones, NPY acts as a neurotransmitter. In the sympathetic nervous system, NPY is frequently localized in noradrenergic neurons and apparently functions both as a vasoconstrictor and as a cotransmitter with norepinephrine. The remainder of this section focuses on NPY.
NPY produces a variety of central nervous system effects, including increased feeding (it is one of the most potent orexigenic molecules in the brain), hypotension, hypothermia, respiratory depression, and activation of the hypothalamic-pituitary-adrenal axis. Other effects include vasoconstriction of cerebral blood vessels, positive chronotropic and inotropic actions on the heart, and hypertension. The peptide is a potent renal vasoconstrictor and suppresses renin secretion, but can cause diuresis and natriuresis. Prejunctional neuronal actions include inhibition of transmitter release from sympathetic and parasympathetic nerves. Vascular actions include direct vasoconstriction, potentiation of the action of vasoconstrictors, and inhibition of the action of vasodilators.
The diverse effects of NPY (and PP and PYY) are mediated by four subtypes of NPY receptors designated Y1, Y2, Y4, and Y5. All are Gi protein-coupled receptors linked to mobilization of Ca2+ and inhibition of adenylyl cyclase. Y1 and Y2 receptors are of major importance in the cardiovascular and other peripheral effects of the peptide. Y4 receptors have a high affinity for pancreatic polypeptide and may be a receptor for the pancreatic peptide rather than for NPY. Y5 receptors are found mainly in the central nervous system and may be involved in the control of food intake. They also mediate the activation of the hypothalamic-pituitary-adrenal axis by NPY.
Selective nonpeptide NPY receptor antagonists are now available for research. The first nonpeptide Y1 receptor antagonist, BIBP3226, is also the most thoroughly studied. It has a short half-life in vivo. In animal models, it blocks the vasoconstrictor and pressor responses to NPY. Structurally related Y1 antagonists include BIB03304 and H409/22; the latter has been tested in humans. SR120107A and SR120819A are orally active Y1 antagonists and have a long duration of action. BIIE0246 is the first nonpeptide antagonist selective for the Y2 receptor; it does not cross the blood-brain barrier. Useful Y4antagonists are not available. The Y5 antagonists MK-0557 and S-2367 have been tested in clinical trials for obesity.
These drugs have been useful in analyzing the role of NPY in cardiovascular regulation. It now appears that the peptide is not important in the regulation of hemodynamics under normal resting conditions but may be of increased importance in cardiovascular disorders including hypertension and heart failure. Other studies have implicated NPY in eating disorders, obesity, alcoholism, anxiety, depression, epilepsy, pain, cancer, and bone physiology. Y1 and particularly Y5 receptor antagonists have potential as antiobesity agents.
![]() UROTENSIN
UROTENSIN
Urotensin II (UII) was originally identified in fish, but isoforms are now known to be present in the human and other mammalian species. Human UII is an 11-amino acid peptide. An eight-amino-acid peptide, UII-related peptide (URP), which is almost identical to the C-terminal of UII has also been identified. Major sites of UII expression in humans include the brain, spinal cord, and kidneys. UII is also present in plasma, and potential sources of this circulating peptide include the heart, lungs, liver, and kidneys. The stimulus to UII release has not been identified but increased blood pressure has been implicated in some studies.
In vitro, UII is a potent constrictor of vascular smooth muscle; its activity depends on the type of blood vessel and the species from which the vessel was obtained. Vasoconstriction occurs primarily in arterial vessels, where UII can be more potent than ET-1, making it the most potent known vasoconstrictor. However, under some conditions, UII may cause vasodilation. In vivo, UII has complex hemodynamic effects, the most prominent being regional vasoconstriction and cardiac depression. In some ways, these effects resemble those produced by ET-1. Nevertheless, the role of the peptide in the normal regulation of vascular tone and blood pressure in humans appears to be minor. In addition to its cardiovascular effects, UII exerts osmoregulatory actions, induces collagen and fibronectin accumulation, modulates the inflammatory response, and inhibits glucose-induced insulin release.
The actions of UII are mediated by a Gq protein-coupled receptor referred to as the UT receptor. UT receptors are widely distributed in the brain, spinal cord, heart, vascular smooth muscle, skeletal muscle, and pancreas. They are located at the cell surface, but specific UII-binding sites have also been observed in heart and brain cell nuclei. Some effects of the peptide including vasoconstriction are mediated by the phospholipase C, inositol trisphosphate, diacylglycerol signal transduction pathway.
Although UII appears to play only a minor role in health, evidence is accumulating that it is involved in cardiovascular and other diseases. In particular, it has been reported that plasma UII levels are increased in hypertension, heart failure, atherosclerosis, diabetes mellitus, and renal failure. For this reason, the development of UII receptor antagonists is of considerable interest. Urantide (“urotensin antagonist peptide”) is a penicillamine-substituted derivative of UII. A nonpeptide antagonist, palosuran, may benefit diabetic patients with renal disease but lacks potency. Recently, more potent UII antagonists have become available. Two are in phase 1 clinical trials, one for the treatment of diabetic nephropathy (EP2439193), the other (SB1440115) for the treatment of asthma.
SUMMARY Drugs That Interact with Vasoactive Peptide Systems
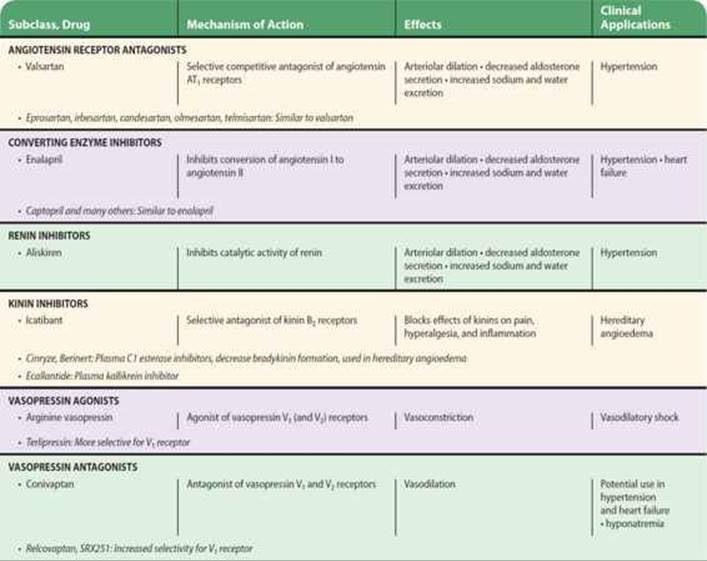
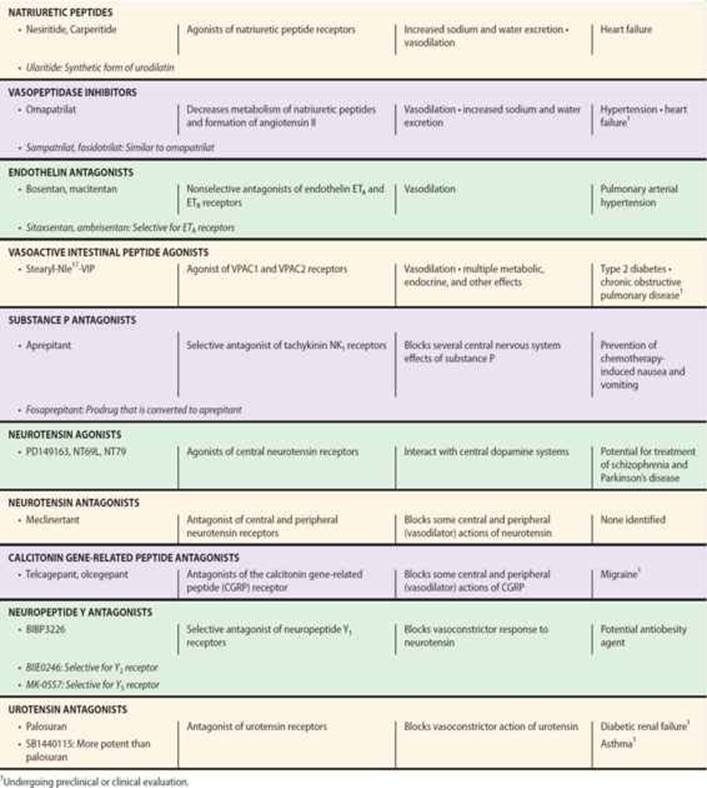
PREPARATIONS AVAILABLE


REFERENCES
Angiotensin
Balakumar P, Jagadeesh G: Cardiovascular and renal pathologic implications of prorenin, renin, and the (pro)renin receptor: Promising young players from the old renin-angiotensin-aldosterone system. J Cardiovasc Pharmacol 2010;56:570.
Castrop H et al: Physiology of kidney renin. Physiol Rev 2010;90:607.
Crowley SD, Coffman TM: Recent advances involving the renin-angiotensin system. Exp Cell Res 2012;318:1049.
Damkjaer M et al: Renal renin secretion as regulator of body fluid homeostasis. Pflugers Archiv Eur J Physiol 2013;465:153.
Friis UG et al: Regulation of renin secretion by renal juxtaglomerular cells. Eur J Physiol 2013;465:25.
Gilbert CJ et al: No increase in adverse events during aliskiren use among ontario patients receiving angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers. Can J Cardiol 2013;29:586.
Harel Z et al: The effect of combination treatment with aliskiren and blockers of the renin-angiotensin system on hyperkalaemia and acute kidney injury: Systematic review and meta-analysis. BMJ 2012;344:e42.
Kim SM, Briggs JP, Schnermann J: Convergence of major physiological stimuli for renin release on the Gs-alpha/cyclic adenosine monophosphate signaling pathway. Clin Exp Nephrol 2012;16:17.
Kurtz A: Renin release: Sites, mechanisms, and control. Annu Rev Physiol 2011;73:377.
McMurray JJ et al: Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med 2010;362:1477.
Nguyen G: Renin, (pro)renin and receptor: An update. Clin Sci (Lond) 2011;120:169.
Sihn G et al: Physiology of the (pro)renin receptor: Wnt of change? Kidney Int 2010;78:246.
Zheng Z et al: A systematic review and meta-analysis of aliskiren and angiotensin receptor blockers in the management of essential hypertension. J Renin Angiotensin Aldosterone Syst 2011;12:102.
Kinins
Cicardi M et al: Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 2010;363:523.
Cicardi M et al: Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010;363:532.
Costa-Neto CM et al: Participation of kallikrein-kinin system in different pathologies. Int Immunopharmacol 2008;8:135.
Pathak M et al: Structure of plasma and tissue kallikreins. Thromb Haemost 2013;109:423.
Vasopressin
Fujiwara Y et al: The roles of V1a vasopressin receptors in blood pressure homeostasis: A review of studies on V1a receptor knockout mice. Clin Exp Nephrol 2012;16:30.
Koshimizu TA et al: Vasopressin V1a and V1b receptors: From molecules to physiological systems. Physiol Rev 2012;92:1813.
Manning M et al: Peptide and non-peptide agonists and antagonists for the vasopressin and oxytocin V1a, V1b, V2 and OT receptors: Research tools and potential therapeutic agents. Prog Brain Res 2008;170:473.
Maybauer MO et al: Physiology of the vasopressin receptors. Best Pract Res Clin Anaesthesiol 2008;22:253.
Natriuretic Peptides
Boerrigter G, Burnett JC Jr: Natriuretic peptides renal protective after all? J Am Coll Cardiol 2011;58:904.
Federico C: Natriuretic peptide system and cardiovascular disease. Heart Views 2010;11:1.
Rubattu S et al: Atrial natriuretic peptide and regulation of vascular function in hypertension and heart failure: Implications for novel therapeutic strategies. J Hypertension 2013;31:1061.
Saito Y: Roles of atrial natriuretic peptide and its therapeutic use. J Cardiol 2010;56:262.
Vogel MW, Chen HH: Novel natriuretic peptides: New compounds and new approaches. Curr Heart Fail Rep 2011;8:22.
Endothelins
Cartin-Ceba R et al: Safety and efficacy of ambrisentan for the therapy of portopulmonary hypertension. Chest 2011;139:109.
Kasimay O et al: Diet-supported aerobic exercise reduces blood endothelin-1 and nitric oxide levels in individuals with impaired glucose tolerance. J Clin Lipidol 2010;4:427.
Kawanabe Y, Nauli SM: Endothelin. Cell Mol Life Sci 2011;68:195.
Pulido T et al: Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369:809.
Sidharta PN, van Giersbergen PL, Dingemanse J: Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects. J Clin Pharmacol 2013;53:1131.
Sitbon O, Morrell N: Pathways in pulmonary arterial hypertension: The future is here. Eur Respir Rev 2012;21:321.
Vasoactive Intestinal Peptide
White CM et al: Therapeutic potential of vasoactive intestinal peptide and its receptors in neurological disorders. CNS Neurol Disord Drug Targets 2010;9:661.
Substance P
Curran MP, Robinson DM: Aprepitant: A review of its use in the prevention of nausea and vomiting. Drugs 2009;69:1853.
Munoz M, Covenas R: Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013;48C:1.
Neurotensin
Boules M et al: Diverse roles of neurotensin agonists in the central nervous system. Front Endocrinol 2013;4:36.
Ferraro L et al: Emerging evidence for neurotensin receptor 1 antagonists as novel pharmaceutics in neurodegenerative disorders. Mini Rev Med Chem 2009;9:1429.
Hwang JI et al: Phylogenetic history, pharmacological features, and signal transduction of neurotensin receptors in vertebrates. Ann N Y Acad Sci 2009;1163:169.
Calcitonin Gene-Related Peptide
Durham PL, Vause CV: Calcitonin gene-related peptide (CGRP) receptor antagonists in the treatment of migraine. CNS Drugs 2010;24:539.
Moore EL, Salvatore CA: Targeting a family B GPCR/RAMP receptor complex: CGRP receptor antagonists and migraine. Br J Pharmacol 2012;166:66.
Walker CS et al: Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol Sci 2010;31:476.
Adrenomedullin
Bell D, McDermott BJ: Intermedin (adrenomedullin-2): A novel counter-regulatory peptide in the cardiovascular and renal systems. Br J Pharmacol 2008;153(Suppl 1):S247.
Nishikimi T et al: Adrenomedullin in cardiovascular disease: A useful biomarker, its pathological roles and therapeutic application. Curr Protein Pept Sci 2013;14:256.
Woolley MJ, Conner AC: Comparing the molecular pharmacology of CGRP and adrenomedullin. Curr Protein Pept Sci 2013;14:358.
Neuropeptide Y
Brothers SP, Wahlestedt C: Therapeutic potential of neuropeptide Y (NPY) receptor ligands. EMBO Mol Med 2010;2:429.
Pedragosa-Badia X, Stichel J, Beck-Sickinger AG: Neuropeptide Y receptors: How to get subtype selectivity. Front Endocrinol 2013;4:5.
Thorsell A: Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp Biol Med 2010;235:1163.
Urotensin
Chatenet D et al: Update on the urotensinergic system: New trends in receptor localization, activation, and drug design. Front Endocrinol 2013;3:1.
Cheriyan J et al: The effects of urotensin II and urantide on forearm blood flow and systemic haemodynamics in humans. Br J Clin Pharmacol 2009;68:518.
Vaudry H et al: Urotensin II, from fish to human. Ann N Y Acad Sci 2010;1200:53.
General
Hoyer D, Bartfai T: Neuropeptides and neuropeptide receptors: Drug targets, and peptide and non-peptide ligands: A tribute to Prof. Dieter Seebach. Chem Biodivers 2012;9:2367.
Paulis L, Unger T: Novel therapeutic targets for hypertension. Nat Rev Cardiol 2010;7:431.
Takahashi K et al: The renin-angiotensin system, adrenomedullins and urotensin II in the kidney: Possible renoprotection via the kidney peptide systems. Peptides 2009;30:1575.
CASE STUDY ANSWER
Enalapril lowers blood pressure by blocking the conversion of angiotensin I to angiotensin II (ANG II). Since converting enzyme also inactivates bradykinin, enalapril increases bradykinin levels, and this is responsible for adverse side effects such as cough and angioedema. This problem might be avoided by using a renin inhibitor, eg, aliskiren, or an ANG II receptor antagonist, eg, losartan, instead of an ACE inhibitor, to block the renin-angiotensin system.