The role of COX enzymes in the actions of NSAIDs
NSAIDs
Mechanisms of action
Classification of NSAIDs
Actions and effects of non-selective NSAIDs
Pharmacokinetics
Unwanted effects
COX-2-selective inhibitors
Paracetamol
Indications for using NSAIDs
The role of COX enzymes in the actions of NSAIDs
The major therapeutic and unwanted actions of non-steroidal anti-inflammatory drugs (NSAIDs) are achieved by inhibition of cyclo-oxygenase (COX) enzymes. COX enzymes are essential in the production of the prostanoids (prostaglandins, prostacyclins and thromboxanes), the most active of which are generated from the 20-carbon, polyunsaturated (ω-6) fatty acid arachidonic acid (Fig. 29.1). Arachidonic acid can also be converted via the 5-lipoxygenase pathway to leukotrienes, some of the actions of which are blocked by leukotriene receptor antagonists (Ch. 12). These classic eicosanoid families are local mediators that are generally synthesised and catabolised close to their site of action, and have numerous physiological actions. Arachidonic acid and other essential fatty acids are also the precursors of novel families of lipid mediators including lipoxins, resolvins, protectins, hepoxilins and endocannabinoids.

FIG. 29.1 The arachidonic acid cascade of eicosanoid synthesis.
Arachidonic acid liberated from membrane phospholipids can be utilised by cyclo-oxygenases (COX-1, COX-2, COX-3) to form prostanoids (prostaglandins, prostacyclin and thromboxane) or by the 5-lipoxygenase pathway to form leukotrienes. The types and amounts of these eicosanoid products that are generated depend on the relative expression of the COX isozymes, 5-lipoxygenase and their respective downstream synthases in different cell types. After release from the cell, the eicosanoids have a multitude of actions via their selective G-protein-coupled receptors on the surface of target cells, such as bronchial, uterine and vascular smooth muscle cells, endothelial cells, platelets and leucocytes (Table 29.2). FLAP, five-lipoxygenase activating protein; 5-HPETE, 5-hydroperoxyeicosatetraenoic acid; LT, leukotriene; PG, prostaglandin; TX, thromboxane.
Arachidonic acid is mostly derived from dietary linoleic acid, which is found in vegetable oils such as sunflower oil. Linoleic acid is converted in the liver in several steps to arachidonic acid, which is then incorporated into glycerophospholipids in cell membranes. Arachidonic acid is released from membrane phospholipids by lipases such as phospholipase A2. In the COX pathways, the initial products of arachidonic acid metabolism are unstable intermediates known as cyclic endoperoxides. These are converted by cell-specific synthases and isomerases to various receptor-active prostanoids (Fig. 29.1). The products of the COX pathway therefore differ among various tissues depending upon the synthases present, generating a diverse range of actions tailored to the individual requirements of each cell type. Most cell types can form different prostanoids simultaneously and in different quantities, with the pattern of production modulated by regulatory influences on the cell.
There are three COX isoenzymes: COX-1, COX-2 and COX-3. COX-1 is constitutively expressed in the endoplasmic reticulum of most cell types. Prostanoids generated via the COX-1 enzyme are produced in small amounts by many cells in the resting state and contribute to the regulation of several homeostatic processes such as renal and gastric blood flow, gastric cytoprotection and platelet aggregation (Table 29.1). COX-2 is present in greater concentrations in the nuclear envelope than in the endoplasmic reticulum. It is found in many cells such as endothelial cells, macrophages, synovial fibroblasts, mast cells, chondrocytes and osteoblasts. Its expression in these cells is induced by cytokines and other inflammatory stimuli, so that COX-2-derived prostanoids are formed in large amounts in response to inflammation, pain and fever. However, the concept that there is only a pathological role for inducible COX-2-derived prostanoids and a ‘housekeeping’ physiological role for the constitutive COX-1 enzyme is now recognised as too simplistic. In many organs, particularly the kidney, central nervous system (CNS), cardiovascular system and reproductive system, there is no clear separation between the functions of COX-1 and COX-2. Either isoform may be involved in the production of ‘physiological’ or ‘pathological’ prostanoids (Table 29.1).
Table 29.1
Some biological roles of cyclo-oxygenase (COX) 1 and 2
|
COX-1 homeostatic roles |
COX-2 homeostatic roles |
|
Gastrointestinal protection |
Renal function |
|
Platelet aggregation |
CNS function |
|
Blood flow regulation |
Tissue repair and healing (including gastrointestinal) |
|
CNS function |
Reproduction |
|
Uterine contraction |
|
|
Blood vessel dilation |
|
|
Pancreas |
|
|
Inhibition of platelet aggregation |
|
|
Airways |
|
|
COX-1 pathological roles |
COX-2 pathological roles |
|
(Possible involvement in inflammation) |
Inflammation |
|
Chronic pain |
Chronic pain |
|
Raised blood pressure |
Fever |
|
Blood vessel permeability |
|
|
Reproduction |
|
|
Alzheimer's disease |
|
|
Angiogenesis, inhibition of apoptosis |
|
|
Tumour cell growth |
COX-3 is a splice variant of COX-1 that retains the entire COX-1 transcript and has an additional conserved intronic sequence. It is mainly expressed in the brain and spinal cord and to a lesser extent in the heart, endothelial cells and monocytes. COX-3 may have a role in pain perception, but its precise functions are uncertain.
The actions of prostaglandins and thromboxanes depend upon the circumstances and site of their formation, and whether they are formed in excessive amounts. For example, prostaglandin E2 (PGE2) is generated in low physiological amounts by COX-1 in gastric mucosa where it is important for maintaining mucosal integrity by a variety of mechanisms, including bicarbonate and mucus production and maintaining mucosal blood flow. In contrast, damage to many tissues leads to increased PGE2 synthesis via enhanced COX-2 expression. This contributes to inflammation and pain by vasodilation, increased vascular permeability and sensitisation of pain fibre nerve endings to the nociceptive action of bradykinin, serotonin and other mediators (Ch. 19). However, in the later stages of repair following tissue damage, COX-2-derived prostanoids may contribute to the processes of wound healing.
Prostanoids act via five main classes of G-protein-coupled receptors on cell surfaces (Fig. 29.1). Some of the actions of prostaglandins are shown in Table 29.2.
Table 29.2
Main biological actions of the eicosanoids
|
Tissue |
Effect |
Eicosanoid |
|
Platelets |
Increased aggregation |
TXA2 |
|
Decreased aggregation |
PGI2, PGD2 |
|
|
Vascular smooth muscle |
Vasodilation |
PGI2, PGE2, PGD2 |
|
Vasoconstriction |
TXA2, LTC4, LTD4 |
|
|
Other smooth muscle |
Bronchodilation |
PGE2 |
|
Bronchoconstriction |
LTC4, LTD4, LTE4, PGD2, PGF2, TXA2 |
|
|
Gastrointestinal tract (contraction/relaxation, depends on muscle type) |
PGF2, PGE2, PGI2, PGD2 |
|
|
Uterine contraction |
PGE2, PGF2α |
|
|
Vascular endothelium |
Increased permeability |
LTC4, LTD4, LTB4 |
|
Potentiates histamine/bradykinin |
PGE2, PGI2 |
|
|
Leucocytes |
Chemotaxis of neutrophils, monocytes, lymphocytes |
LTB4 |
|
Chemotaxis of eosinophils, basophils |
LTE4, PGD2 |
|
|
Gastrointestinal mucosa |
Reduced acid secretion |
PGE2, PGI2 |
|
Increased mucus secretion |
PGE2 |
|
|
Increased blood flow |
PGE2, PGI2 |
|
|
Nervous system |
Inhibition of noradrenaline release |
PGD2, PGE2, PGI2 |
|
Endogenous pyrogen in hypothalamus |
PGE2 |
|
|
Sedation, sleep |
PGD2 |
|
|
Endocrine/metabolic |
Secretion of ACTH, GH, prolactin, gonadotrophins |
PGE2 |
|
Inhibition of lipolysis |
PGE2 |
|
|
Kidney |
Increased renal blood flow |
PGE2, PGI2 |
|
Antagonism of ADH |
PGE2 |
|
|
Renin release |
PGI2, PGE2, PGD2 |
|
|
Pain |
Potentiates pain through bradykinin, serotonin |
PGE2, PGD2 |
Only the main eicosanoids are shown. Inhibition of COX isoenzymes by NSAIDs reduces the synthesis only of prostanoids. Synthesis of leukotrienes is reduced by 5-lipoxygenase inhibitors (zileuton, not available in UK). Antagonists of the cysteinyl-leukotriene (LTC4, LTD4, LTE4) receptor 1 are used in asthma prophylaxis (Ch. 12).
ACTH, adrenocorticotrophic hormone (corticotropin); ADH, antidiuretic hormone (vasopressin); GH, growth hormone; LT, leukotriene; PG, prostaglandin; TX, thromboxane.
The second route for arachidonic acid metabolism is via the 5-lipoxygenase pathway to produce leukotrienes (Fig. 29.1). These are also involved in the inflammatory process by enhancing vascular permeability and smooth muscle contraction (particularly the cysteinyl-leukotrienes LTC4, LTD4 and LTE4; see also Ch. 12) and through chemotactic attraction of leucocytes (particularly LTB4) (Table 29.2).
NSAIDs
NSAIDs have three major therapeutic activities: anti-inflammatory, analgesic and antipyretic.
Mechanisms Of Action
All NSAIDs share a common mode of action by inhibition of the COX isoenzymes (Table 29.3). Most NSAIDs bind reversibly to the site in the COX enzymes that accepts arachidonic acid, but the irreversible inactivation produced by aspirin (acetylsalicylic acid) involves acetylation of a serine residue in the enzyme. This mechanism of action is important in the use of aspirin as an antiplatelet drug (see below and Ch. 11).
Table 29.3
Selectivity of some NSAIDs for inhibition of COX-1 compared with COX-2

Different NSAIDs inhibit the two main isoenzymes (COX-1 and COX-2) to varying extents. The degree of COX selectivity of each NSAID will also depend on the dosage used. The clinical responses and unwanted effect profiles of individual NSAIDs reflect the ability of the drug to inhibit the diverse biological actions of each isoenzyme (Tables 29.1 and 29.2). Highly selective inhibitors of COX-2 (coxibs) have been developed with the aim of reducing the unwanted effects associated with concomitant inhibition of COX-1 by the non-selective NSAIDs (see below). Neither the non-selective NSAIDs nor the selective COX-2 inhibitors directly affect the production of leukotrienes by 5-lipoxygenase. However, PGE2 normally inhibits leukotriene synthesis and reduced PGE2 generation as a consequence of NSAID use may increase leukotriene synthesis.
NSAIDs have additional anti-inflammatory effects that appear to be independent of COX inhibition. These effects may be mediated by actions on peroxisome proliferator-activated receptors (PPARs), particularly PPAR-γ. PPARs have key roles in modulating immune responses by suppressing transcription of pro-inflammatory genes, such as those encoding tumour necrosis factor α (TNFα), interleukin-1 (IL-1) and inducible nitric oxide synthase (iNOS) in macrophages. NSAIDs also have complex effects on lymphocytes, including inhibition of T-cell activation and increased T-cell apoptosis, which may reflect PPAR-mediated actions. NSAIDs may also modulate inflammatory gene transcription via actions on the nuclear transcription factor nuclear factor κB (NF-κB) and on other cell-signalling proteins including mitogen-activated protein (MAP) kinases. Some of these COX-independent actions only occur in vitro at NSAID concentrations higher than those required for anti-inflammatory activity or analgesia in vivo, and their clinical relevance remains to be determined.
Classification Of NSAIDs
Table 29.3 shows the principal chemical types of NSAIDs with an indication of their selectivity in inhibiting COX-1 or COX-2. Many classical NSAIDs produce greater inhibition of COX-1 than of COX-2, although at clinical doses this selectivity may not be apparent and both isoenzymes will be effectively inhibited. The coxibs are highly selective inhibitors of COX-2 at clinical doses.
Actions And Effects Of Non-Selective NSAIDs
Some of the properties and actions of a selection of commonly used analgesic drugs are compared in Table 29.4.
Table 29.4
Properties of some commonly used analgesic drugs
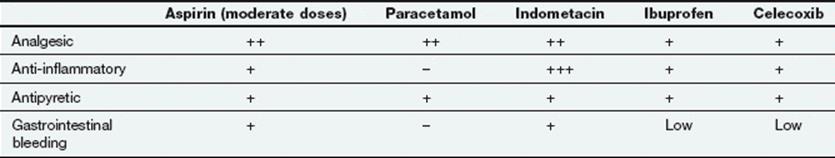
Analgesic effect
The analgesic action of NSAIDs is in part a peripheral action at the site of pain and is most effective when the pain has an inflammatory origin (see Ch. 19). It is achieved predominantly through inhibition of COX-2-derived prostaglandins in inflamed or injured tissues. The main pain-inducing action of prostanoids is at sensory nerve endings on first-order neurons in peripheral tissues. PGE2 enhances the ability of several mediators (serotonin, substance P, bradykinin) to stimulate Aδ and C nociceptive fibres. There is also a CNS component to the analgesic action of NSAIDs which is due to inhibition of COX isoenzymes and reduction of prostaglandin production within the CNS pain pathways. COX inhibition decreases PGE2 production in the dorsal horn of the spinal cord, which inhibits neurotransmitter release and reduces the excitability of second-order neurons in the pain relay pathway. The analgesic action of NSAIDs is apparent after the first dose, but does not reach its maximal effect until about 1 week with repeated dosing.
Anti-inflammatory effect
Inhibition of vasodilation and oedema is partly related to a reduction in peripheral COX-2-generated prostaglandin synthesis. However, NSAIDs also affect several other inflammatory processes unrelated to their effects on prostaglandins. For example, they probably reduce harmful superoxide free radical generation by neutrophils. They may also uncouple G-protein-regulated processes in the cell membrane of inflammatory cells, which reduces their responsiveness to a variety of agonists released by damaged tissues. The anti-inflammatory effects of NSAIDs develop gradually over about 3 weeks.
Antipyretic effect
Fever is reduced through inhibition of hypothalamic COX-2. Circulating pyrogens such as interleukin-1 enhance PGE2 production in the hypothalamus, which depresses the response of temperature-sensitive neurons. NSAIDs do not affect normal body temperature.
Reduction of platelet aggregation
This action is mediated by inhibition of the synthesis of thromboxane A2 (TXA2), a potent platelet-aggregating agent, by platelet COX-1 (Ch. 11). This effect is most marked for aspirin, because it has an irreversible action on COX, and platelets are unable to synthesise more enzyme during their life span. The reversible action of most other NSAIDs produces weaker platelet inhibition (with the exception of naproxen), and selective COX-2 inhibitors do not inhibit platelet aggregation (Table 29.3).
Pharmacokinetics
Most NSAIDs are weak acids that undergo some absorption from the stomach due to pH partitioning (Ch. 2). This explains the relatively high drug concentration in cells of the gastric mucosa. However, the majority of the drug is absorbed via the larger surface area of the small bowel.
Absorption of NSAIDs from the gut is usually fairly rapid from conventional formulations. Some NSAIDs with short half-lives, such as diclofenac, are available as modified-release formulations to prolong their duration of action. Certain NSAIDs can be given by intramuscular or intravenous injection for rapid onset of postoperative analgesia (such as ketorolac), or rectally to achieve a prolonged action (such as diclofenac and ketoprofen). Transcutaneous delivery of several NSAIDs, usually as a gel formulation, was introduced with the intention of providing high local drug concentrations while attempting to minimise systemic unwanted effects. However, once the drug has penetrated the skin it is widely distributed, and this route has little advantage for reducing systemic toxicity.
Most NSAIDs undergo hepatic metabolism to inactive compounds and differ widely in their elimination half-lives. Short-acting drugs require frequent dosing to maintain continuous therapeutic effect, although synovial fluid concentrations in joint disease fluctuate less than the plasma concentrations. Piroxicam undergoes enterohepatic cycling, which contributes to its long half-life.
Aspirin (acetylsalicylic acid) is initially converted to an active metabolite, salicylic acid, and finally inactivated by conjugation with glycine and, to some extent, glucuronic acid. Conjugation with glycine is saturable at higher doses and the metabolism of salicylate then changes from first-order to zero-order elimination kinetics (Ch. 2). This has important implications for aspirin overdose (Ch. 53).
Unwanted Effects
Most unwanted effects arise in part from the inhibition of prostaglandin synthesis throughout the body. They are usually dose-related.
Gastrointestinal effects
![]() Nausea, dyspepsia, gastric irritation and gastric ulceration are the most frequent unwanted effects. They occur principally as a result of inhibition of mucosal production of COX-1-generated PGE2 and PGI2, although inhibition of COX-2 may interfere with some aspects of tissue healing. PGE2 has several actions that confer cytoprotection in the stomach (Ch. 33). There are many mechanisms by which NSAIDs cause gastric irritation, as follows.
Nausea, dyspepsia, gastric irritation and gastric ulceration are the most frequent unwanted effects. They occur principally as a result of inhibition of mucosal production of COX-1-generated PGE2 and PGI2, although inhibition of COX-2 may interfere with some aspects of tissue healing. PGE2 has several actions that confer cytoprotection in the stomach (Ch. 33). There are many mechanisms by which NSAIDs cause gastric irritation, as follows.
![]() Mucus secretion and bicarbonate secretion are reduced and acid secretion is increased as a result of inhibition of prostaglandin synthesis
Mucus secretion and bicarbonate secretion are reduced and acid secretion is increased as a result of inhibition of prostaglandin synthesis
![]() Inhibition of prostaglandin synthesis also reduces mucosal blood flow, which probably enhances cytotoxicity by producing tissue hypoxia and enhanced local generation of free radicals.
Inhibition of prostaglandin synthesis also reduces mucosal blood flow, which probably enhances cytotoxicity by producing tissue hypoxia and enhanced local generation of free radicals.
![]() The mucus gel layer is rendered less hydrophobic due to the acidic nature of NSAIDs and their local concentration in gastric mucosal cells; this reduces the barrier effect of the surface layer.
The mucus gel layer is rendered less hydrophobic due to the acidic nature of NSAIDs and their local concentration in gastric mucosal cells; this reduces the barrier effect of the surface layer.
![]() Uncoupling of cellular oxidative phosphorylation by NSAIDs increases mucosal cell permeability, with consequent back-diffusion of H+ ions, which are trapped in the mucosal epithelium and lead to cytotoxicity.
Uncoupling of cellular oxidative phosphorylation by NSAIDs increases mucosal cell permeability, with consequent back-diffusion of H+ ions, which are trapped in the mucosal epithelium and lead to cytotoxicity.
NSAIDs accumulate within gastric mucosal cells by direct absorption of the drug from the gastric lumen and also by systemic delivery of the drug to the mucosa. Consequently, rectal or transdermal administration or the use of a prodrug may reduce, but will not eliminate, the risk of gastric damage. Occult blood loss from the bowel is increased during regular treatment with NSAIDs and the risk of overt gastrointestinal bleeding is greater. The highest risk of gastrointestinal bleeding is with piroxicam and ketoprofen, while indometacin, diclofenac and naproxen carry an intermediate risk. Ibuprofen has the lowest risk. Management of NSAID-induced gastric damage is considered in Chapter 33.
![]() Exacerbation of inflammatory bowel disease (Ch. 34).
Exacerbation of inflammatory bowel disease (Ch. 34).
![]() Lower gastrointestinal bleeding or perforation.
Lower gastrointestinal bleeding or perforation.
![]() Local irritation and bleeding from rectal administration.
Local irritation and bleeding from rectal administration.
Renal effects
![]() NSAIDs can produce a reversible decline in renal function, with a rise in serum creatinine. Inhibition of prostaglandins (PGE2, PGI2) generated by both COX-1 and COX-2 reduces renal medullary blood flow (Ch. 14). An effect of NSAIDs on renal function is more likely if there is underlying chronic kidney disease (as is often the case in the elderly). There is also an increased risk in the presence of heart failure or cirrhosis, conditions that are associated with reduced effective circulating blood volume, when prostaglandins play a greater role in the maintenance of renal blood flow. NSAIDs are the second most common cause of drug-induced acute kidney injury after aminoglycoside antimicrobials, and this most often occurs during the first few weeks of treatment.
NSAIDs can produce a reversible decline in renal function, with a rise in serum creatinine. Inhibition of prostaglandins (PGE2, PGI2) generated by both COX-1 and COX-2 reduces renal medullary blood flow (Ch. 14). An effect of NSAIDs on renal function is more likely if there is underlying chronic kidney disease (as is often the case in the elderly). There is also an increased risk in the presence of heart failure or cirrhosis, conditions that are associated with reduced effective circulating blood volume, when prostaglandins play a greater role in the maintenance of renal blood flow. NSAIDs are the second most common cause of drug-induced acute kidney injury after aminoglycoside antimicrobials, and this most often occurs during the first few weeks of treatment.
![]() NSAIDs can produce renal salt and water retention even when renal function is normal. Reduced prostaglandin synthesis in the ascending limb of the loop of Henle increases expression of the Na+/K+/2Cl– co-transporter complex, and prostaglandins antagonise the action of vasopressin (antidiuretic hormone, ADH; see Ch. 14). Water retention due to the unopposed action of ADH may exceed retention of Na+, resulting in dilutional hyponatraemia. Salt and water retention produced by NSAIDs can exacerbate heart failure and raises blood pressure by an average of 3–5 mmHg. In addition, the efficacy of drug treatments for these conditions (e.g. diuretics, angiotensin-converting enzyme inhibitors, β-adrenoceptor antagonists) is blunted by NSAIDs.
NSAIDs can produce renal salt and water retention even when renal function is normal. Reduced prostaglandin synthesis in the ascending limb of the loop of Henle increases expression of the Na+/K+/2Cl– co-transporter complex, and prostaglandins antagonise the action of vasopressin (antidiuretic hormone, ADH; see Ch. 14). Water retention due to the unopposed action of ADH may exceed retention of Na+, resulting in dilutional hyponatraemia. Salt and water retention produced by NSAIDs can exacerbate heart failure and raises blood pressure by an average of 3–5 mmHg. In addition, the efficacy of drug treatments for these conditions (e.g. diuretics, angiotensin-converting enzyme inhibitors, β-adrenoceptor antagonists) is blunted by NSAIDs.
![]() Suppression of prostaglandin-mediated renin secretion by NSAIDs can lead to hypoaldosteronism and hyperkalaemia.
Suppression of prostaglandin-mediated renin secretion by NSAIDs can lead to hypoaldosteronism and hyperkalaemia.
![]() Acute interstitial nephritis is a less common cause of renal impairment and can occur with any NSAID. It often becomes apparent after many months of NSAID use.
Acute interstitial nephritis is a less common cause of renal impairment and can occur with any NSAID. It often becomes apparent after many months of NSAID use.
Hypersensitivity
Hypersensitivity reactions occasionally produce asthma, urticaria, angioedema and rhinitis. People with nasal polyps and known allergic disorders appear to be most susceptible. NSAIDs can also precipitate ‘pseudo-allergic’ asthma in a subgroup of people with asthma, through inhibition of COX-1-generated PGE2 production in the lung. PGE2 has an inhibitory effect on leukotriene synthesis and mast cell degranulation. A reduction in PGE2 leads to an increased synthesis of bronchoconstrictor cysteinyl-leukotrienes (LTC4, LTD4, LTE4) (Ch. 12). It is suggested that as many as one in five people with asthma may be sensitive to NSAIDs.
Other unwanted effects
Other unwanted effects are unrelated to prostaglandin inhibition and are sometimes specific for individual compounds.
![]() CNS unwanted effects such as headache, dizziness, drowsiness, insomnia and confusion, particularly in the elderly.
CNS unwanted effects such as headache, dizziness, drowsiness, insomnia and confusion, particularly in the elderly.
![]() Rashes.
Rashes.
![]() Tinnitus in toxic doses; overdose of aspirin can be particularly hazardous (Ch. 53).
Tinnitus in toxic doses; overdose of aspirin can be particularly hazardous (Ch. 53).
![]() Aspirin can cause Reye's syndrome in children, a rare condition producing acute encephalopathy and fatty degeneration of the liver. Aspirin should be avoided in children under the age of 12 years.
Aspirin can cause Reye's syndrome in children, a rare condition producing acute encephalopathy and fatty degeneration of the liver. Aspirin should be avoided in children under the age of 12 years.
![]() The risk of myocardial infarction and stroke is increased by most NSAIDs, especially in people with known ischaemic heart disease or at high risk of a cardiovascular event. Naproxen may be associated with a lower risk than other NSAIDs.
The risk of myocardial infarction and stroke is increased by most NSAIDs, especially in people with known ischaemic heart disease or at high risk of a cardiovascular event. Naproxen may be associated with a lower risk than other NSAIDs.
COX-2-Selective Inhibitors
Examples
celecoxib, etoricoxib, parecoxib
Mechanism of action
Selective COX-2 inhibitors have less inhibitory action on COX-1, but the degree of selectivity for COX-2 varies among the drugs in this class. Selective COX-2 inhibitors have anti-inflammatory actions similar to conventional non-selective NSAIDs, but there is some evidence that they may be less effective analgesics. This may possibly be due to less inhibition of COX-3 in the brain and spinal cord. Selective COX-2 inhibitors have little direct effect on platelet TXA2 production and do not impair platelet aggregation; however, they suppress the production of the anti-aggregatory and vasodilator PGI2 by blood vessels, which may allow TXA2 to exert unopposed aggregatory effects on platelets. Selective COX-2 inhibitors also interact with PPARs and impair macrophage activity and T-cell-mediated immune responses (see above).
Pharmacokinetics
Celecoxib and etoricoxib are well absorbed from the gut. They are eliminated by hepatic metabolism. The half-life of celecoxib is 11 h, while that of etoricoxib is longer (25 h). Parecoxib can be given by intramuscular or intravenous injection for control of postoperative pain and has a half-life of 5–9 h.
Unwanted effects
![]() COX-2-selective inhibitors produce fewer upper gastrointestinal unwanted effects, and celecoxib, but not etoricoxib, reduces the risk of ulcers and ulcer complications by up to 50% compared with conventional NSAIDs. However, if low-dose aspirin is taken concurrently for its antiplatelet benefit, this negates the gastrointestinal-sparing benefits of celecoxib.
COX-2-selective inhibitors produce fewer upper gastrointestinal unwanted effects, and celecoxib, but not etoricoxib, reduces the risk of ulcers and ulcer complications by up to 50% compared with conventional NSAIDs. However, if low-dose aspirin is taken concurrently for its antiplatelet benefit, this negates the gastrointestinal-sparing benefits of celecoxib.
![]() Exacerbation of inflammatory bowel disease (Ch. 34).
Exacerbation of inflammatory bowel disease (Ch. 34).
![]() Stomatitis or mouth ulcers.
Stomatitis or mouth ulcers.
![]() Fatigue, influenza-like symptoms.
Fatigue, influenza-like symptoms.
![]() Palpitation.
Palpitation.
![]() Selective COX-2 inhibitors rarely induce asthma attacks in NSAID-sensitive individuals.
Selective COX-2 inhibitors rarely induce asthma attacks in NSAID-sensitive individuals.
![]() Selective COX-2 inhibitors increase the risk of myocardial infarction in people with known ischaemic heart disease or those who are at high risk of a cardiovascular event. The risk is similar to most non-selective NSAIDs.
Selective COX-2 inhibitors increase the risk of myocardial infarction in people with known ischaemic heart disease or those who are at high risk of a cardiovascular event. The risk is similar to most non-selective NSAIDs.
![]() Renal effects, including salt and water retention and acute kidney injury, are similar for selective COX-2 inhibitors and non-selective NSAIDs.
Renal effects, including salt and water retention and acute kidney injury, are similar for selective COX-2 inhibitors and non-selective NSAIDs.
Paracetamol
Mechanism of action
Paracetamol (acetaminophen in the USA) is an analgesic without anti-inflammatory activity and is not an NSAID. It has very little inhibitory effect on COX-1 or COX-2 in peripheral tissues. In the CNS, paracetamol inhibits COX-2 by reducing the availability of an essential co-substrate for the action of COX-2. In peripheral tissues, hydroperoxides generated during the metabolism of arachidonic acid by lipoxygenases may impair the action of paracetamol. There is weak inhibition of COX-3 in the CNS by paracetamol, but this may not be clinically important.
Other effects may contribute to the analgesic action of paracetamol, such as stimulation of spinal serotonergic neurotransmission via 5-HT1A receptors and modulation of cannabinoid (CB1) receptors (see also Ch. 19). Paracetamol is converted to a metabolite that inhibits neuronal reuptake of the endogenous cannabinoid anandamide. Anandamide acts at cannabinoid CB1 receptors and transient receptor potential vanilloid 1 (TRPV1) receptors, and high concentrations stimulate then desensitize the receptors in a similar manner to capsaicin (Ch.19). Lastly, a different metabolite of paracetamol (N-acetyl-p-benzoquinone imine, NAPQI) stimulates transient receptor potential ankyrin 1 (TRPA1) receptors in the spinal cord, which inhibits voltage-gated Ca2+ and Na+ channels in primary sensory neurons.
Pharmacokinetics
Paracetamol is rapidly absorbed from the gut. It is metabolised mainly by conjugation, but the minor metabolite NAPQI is produced by cytochrome P450 in the liver and kidneys. NAPQI is detoxified by a limited supply of glutathione in these organs. In paracetamol overdose, failure to conjugate this potentially toxic metabolite can lead to liver and renal damage (Ch. 53).
Unwanted effects
![]() Paracetamol is usually well tolerated, and because it does not inhibit peripheral prostaglandin synthesis it does not cause problems with homeostatic functions of prostanoids, for example gastrointestinal disturbances.
Paracetamol is usually well tolerated, and because it does not inhibit peripheral prostaglandin synthesis it does not cause problems with homeostatic functions of prostanoids, for example gastrointestinal disturbances.
![]() Hepatic damage and renal failure in overdose (Ch. 53).
Hepatic damage and renal failure in overdose (Ch. 53).
Indications For Using NSAIDs
NSAIDs are useful for pain relief, particularly for:
![]() inflammatory conditions affecting joints, soft tissues, etc.,
inflammatory conditions affecting joints, soft tissues, etc.,
![]() postoperative pain,
postoperative pain,
![]() renal colic,
renal colic,
![]() headache,
headache,
![]() primary dysmenorrhoea; stimulation of the uterus by prostaglandins can be responsible for the pain in this condition.
primary dysmenorrhoea; stimulation of the uterus by prostaglandins can be responsible for the pain in this condition.
About 60% of people will respond to any one NSAID, but those who fail to respond to one may derive benefit from another. Adequate time must be allowed for the full analgesic or anti-inflammatory effect to develop (see above). The initial choice of NSAID is mainly determined by unwanted effects, particularly on the stomach and the cardiovascular system. For these reasons, ibuprofen is often the NSAID of first choice.
NSAIDs are also used for other conditions not associated with pain:
![]() as an antipyretic in febrile conditions,
as an antipyretic in febrile conditions,
![]() to achieve closure of a patent ductus arteriosus in a neonate where patency may be inappropriately maintained by prostaglandin production; NSAIDs should not be given to a pregnant mother in the third trimester, to avoid premature closure of the ductus,
to achieve closure of a patent ductus arteriosus in a neonate where patency may be inappropriately maintained by prostaglandin production; NSAIDs should not be given to a pregnant mother in the third trimester, to avoid premature closure of the ductus,
![]() for modest reduction of menstrual blood loss in menorrhagia (excessive blood loss at menstruation),
for modest reduction of menstrual blood loss in menorrhagia (excessive blood loss at menstruation),
![]() for prevention of vascular occlusion by inhibition of platelet aggregation (low-dose aspirin; Ch. 11),
for prevention of vascular occlusion by inhibition of platelet aggregation (low-dose aspirin; Ch. 11),
![]() aspirin and other NSAIDs reduce the risk of developing colorectal cancer, oesophageal and gastric cancer, breast cancer, bladder cancer and adenocarcinoma of the lung by about 30%. Aspirin also reduces the risk of metastasis from adenocarcinoma by 50%. The effect of NSAIDs may depend on inhibition of COX isoenzymes or actions on other nuclear transcription and cell signalling pathways that result in reduced cellular proliferation, migration and angiogenesis and enhanced apoptosis.
aspirin and other NSAIDs reduce the risk of developing colorectal cancer, oesophageal and gastric cancer, breast cancer, bladder cancer and adenocarcinoma of the lung by about 30%. Aspirin also reduces the risk of metastasis from adenocarcinoma by 50%. The effect of NSAIDs may depend on inhibition of COX isoenzymes or actions on other nuclear transcription and cell signalling pathways that result in reduced cellular proliferation, migration and angiogenesis and enhanced apoptosis.
Selective COX-2 inhibitors should be used in preference to standard NSAIDs only in people who are at high risk of developing serious gastrointestinal adverse effects and when an NSAID is clearly indicated as part of the management.
Self-Assessment
True/false questions
1. Three cyclo-oxygenase (COX) isoenzymes can synthesise prostanoids.
2. Gastrointestinal complications are the most common unwanted effects of NSAIDs.
3. COX-2 is not found constitutively in cells.
4. Prostaglandin E2 (PGE2) does not cause pain directly.
5. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit COX-1 and COX-2 isoenzymes with equal potency.
6. Paracetamol is a potent analgesic and anti-inflammatory drug.
7. Aspirin is a suitable analgesic for infants.
8. NSAIDs reduce gastric blood flow.
9. Celecoxib causes a greater incidence of gastrointestinal symptoms than naproxen.
10. Misoprostol reduces the gastric damage caused by diclofenac.
11. Antiplatelet doses of aspirin (75 mg per day) can compromise renal function.
12. The elderly have a greater risk of gastrointestinal adverse events when given NSAIDs.
13. Ibuprofen is an effective first-choice NSAID in severe rheumatoid arthritis.
14. Aspirin is a good choice of analgesic therapy for people with asthma.
15. Celecoxib is an antipyretic.
16. NSAIDs increase the risk of colorectal cancer.
Extended-matching-item questions
Choose the most appropriate NSAID from options A–E to be given initially in the case scenarios 1–3 below.
A Aspirin
B Celecoxib
C Diclofenac plus misoprostol
D Paracetamol
E Indometacin
Case 1. An elderly man with a long history of hypertension, congestive heart failure with oedema, and chronic gastritis, has chronic mild knee pain, due to osteoarthritic changes. The pain is interrupting his sleep.
Case 2. A 38-year-old severely asthmatic woman with a history of nasal polyposis and recurrent dyspepsia, a diagnosis of rheumatoid arthritis and no history of heart disease.
Case 3. A 45-year-old man who recently had a myocardial infarction. He was prescribed an angiotensin-converting enzyme (ACE) inhibitor and simvastatin.
True/false answers
1. True. The isoenzymes are COX-1, COX-2 and COX-3 (a splice variant of COX-1).
2. True. Gastric irritation, ulceration and bleeding caused by inhibition of gastroprotective prostanoid synthesis are common unwanted effects of NSAID use.
3. False. COX-2 is present constitutively in some cells such as in blood vessels, but can be markedly induced in these cell types and others by cytokines and inflammatory stimuli.
4. True. PGE2 generated by COX-2 does not directly stimulate sensory pain fibres but sensitises them to bradykinin and other mediators.
5. False. There is a wide range in the selectivity of NSAIDs for inhibition of COX-1 and COX-2. Their anti-inflammatory potency relates broadly to their potency in inhibiting COX-2, and their unwanted gastrointestinal effects to their potency in inhibiting COX-1.
6. False. Paracetamol is analgesic and antipyretic but has only a weak anti-inflammatory effect. The reasons for this are imperfectly understood but it may act by COX-independent mechanisms on pain pathways within the CNS.
7. False. Aspirin can precipitate Reye's syndrome and should not be used in children under 12 years old.
8. True. Reduced blood flow contributes to the gastric damage caused by NSAIDs; they also inhibit bicarbonate and mucus secretion.
9. False. The COX-2-selective inhibitor celecoxib is associated with fewer gastrointestinal unwanted effects than the non-selective NSAID naproxen, although this difference declines with continued use.
10. True. Misoprostol is a PGE1 analogue, also available in combination formulations with diclofenac or naproxen; its use can limit the risk of gastric damage by restoring gastroprotective prostanoid activity.
11. False. Low doses of aspirin are safe, but long-term use of high doses can result in renal ischaemia, sodium and water retention, papillary necrosis and chronic renal failure.
12. True. Gastrointestinal unwanted effects are particularly common in those over 75 years of age and in those with a history of peptic ulcer.
13. False. Ibuprofen is effective in mild-to-moderate arthritis but other NSAIDs such as indometacin or diclofenac have greater anti-inflammatory potential, although a greater propensity to cause unwanted effects.
14. False. In a subgroup of people with asthma, aspirin and most other non-selective NSAIDs can induce an asthmatic episode through the formation of bronchoconstrictor cysteinyl-leukotrienes (LTC4, LTD4). Selective COX-2 inhibitors have less potential to cause asthma attacks in NSAID-intolerant asthmatics.
15. True. Pyrexia is caused by elevation of PGE2 levels synthesised in the CNS by COX-2.
16. False. Increasing evidence suggests that NSAIDs reduce the risk of many cancers, most probably by inhibiting PGE2 synthesis by COX-2, or by COX-independent pathways.
Extended-matching-item answers
Case 1: Answer D is correct. Paracetamol is a good choice for this man's mild osteoarthritic pain. His hypertension and heart failure mean that he should not be given an NSAID that may result in salt and water retention. Both the COX-2-selective and non-selective NSAIDs contribute to salt and water retention.
Case 2: Answer B is correct. The selective COX-2 inhibitor celecoxib could be prescribed as these are less likely than non-selective NSAIDs such as aspirin or diclofenac to precipitate a hypersensitive asthmatic attack. Paracetamol would not be useful as it has little anti-inflammatory action.
Case 3: Answer A is correct. This man should be given low-dose aspirin (75 mg daily) as it prevents platelet aggregation by inhibiting thromboxane A2 (TXA2) synthesis while having a minimal effect on the production of the vasodilator and anti-aggregatory PGI2 (prostacyclin). This low dose has been shown to reduce the risk of another infarction.
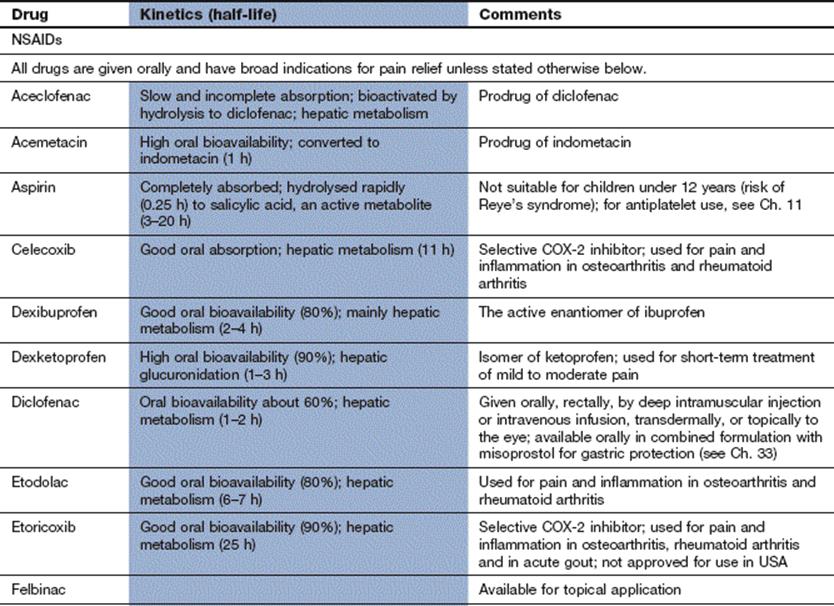
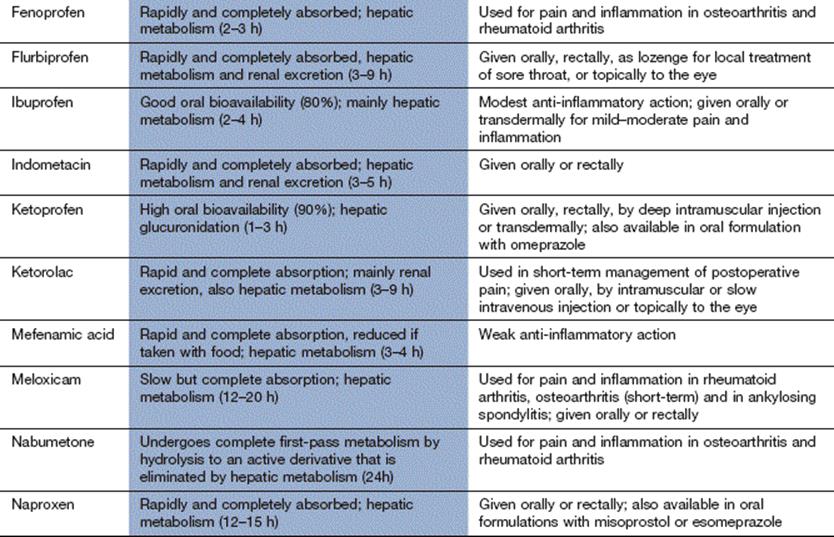
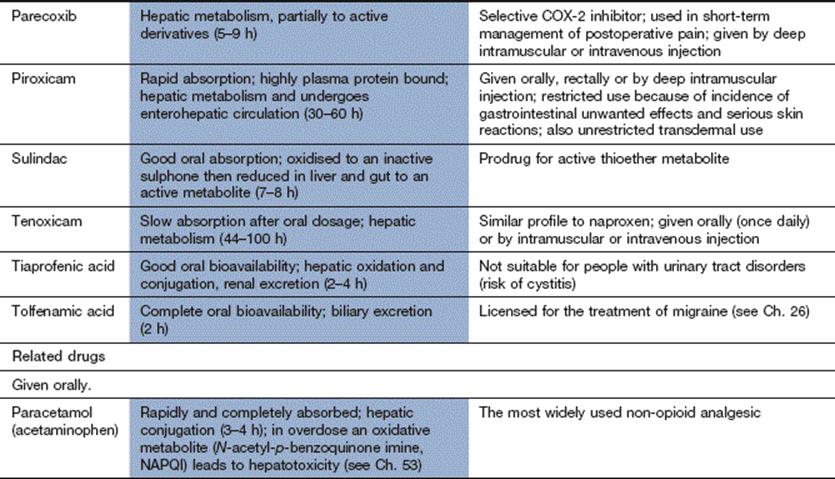
Compendium: non-steroidal anti-inflammatory drugs (NSAIDs) and related drugs
Further reading
Bleumink, GS, Feenstra, J, Sturkenboom, CJM, et al. Nonsteroidal anti-inflammatory drugs and heart failure. Drugs. 2003;63:525–534.
Burian, M, Geisslinger, G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol Ther. 2005;107:139–154.
Epstein, M. Non-steroidal anti-inflammatory drugs and the continuum of renal dysfunction. J Hypertens. 2002;20(suppl 6):S17–S23.
Farooque, SP, Lee, TH. Aspirin-sensitive respiratory disease. Annu Rev Physiol. 2009;71:465–487.
Högestätt, ED, Jönsson, BA, Ermund, A, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280:31405–31412.
James, MW, Hawkey, CJ. Assessment of non-steroidal anti-inflammatory drug (NSAID) damage in the human gastrointestinal tract. Br J Clin Pharmacol. 2003;56:146–155.
Micklewright, R, Linley, SLW, McQuade, C, et al. NSAIDs, gastroprotection and cyclo-oxygenase-II-selective inhibitors. Aliment Pharmacol Ther. 2003;17:321–332.
Paccani, SR, Boncristiano, M, Baldari, CT. Molecular mechanisms underlying suppression of lymphocyte responses by nonsteroidal antiinflammatory drugs. Cell Mol Life Sci. 2003;60:1071–1083.
Parente, L, Perretti, M. Advances in the pathophysiology of constitutive and inducible cyclooxygenases: two enzymes in the spotlight. Biochem Pharmacol. 2003;65:153–159.
Peters-Golden, M, Henderson, WR. Leukotrienes. N Engl J Med. 2007;357:1841–1854.
Serhan, CN, Chiang, N, Van Dyke, TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361.
Stevenson, DD. Aspirin sensitivity and desensitization for asthma and sinusitis. Curr Allergy Asthma Rep. 2009;9:155–163.
Szallasi, A, Cruz, F, Geppetti, P. TRPV1: a therapeutic target for novel analgesic drugs? Trends Mol Med. 2006;12:545–554.
Trelle, S, Reichenbach, S, Wandel, S, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086.
Wang, D, Dubois, RN. Prostaglandins and cancer. Gut. 2006;55:115–122.