MAJOR DRUG CLASSES
Drugs for anxiety (anxiolytics)
Benzodiazepines
Non-benzodiazepine anxiolytics
Benzodiazepine receptor antagonist
Drugs for insomnia
Benzodiazepines
Benzodiazepine receptor agonists
Melatonin receptor agonists
Therapeutic Overview
Anxiety disorders affect 19 million adults in the United States. The term anxiety refers to a pervasive feeling of apprehension and is characterized by diffuse symptoms such as feelings of helplessness, difficulties in concentrating, irritability, and insomnia, as well as somatic symptoms including gastrointestinal disturbances, muscle tension, excessive perspiration, tachypnea, tachycardia, nausea, palpitations, and dry mouth.
Anxiety disorders are chronic and relentless and can progress if not treated. The anxiety disorders include:
• Panic disorder
• Obsessive-compulsive disorder
• Post-traumatic stress disorder
• Social phobia
• Social anxiety disorder
• Generalized anxiety disorder
• Specific phobias
Each disorder has its own distinct features, but they are all bound together by the common theme of excessive, irrational fear of impending doom, loss of control, nervousness, and dread. Depression often accompanies anxiety disorders, and when it does, it should be treated (see Chapter 30).
In many cases anxiety symptoms may be mild and require little or no treatment. However, at other times symptoms may be severe enough to cause considerable distress. When patients exhibit anxiety so debilitating that lifestyle, work, and interpersonal relationships are severely impaired, they may require drug treatment. Although these compounds may be of great benefit, concurrent psychological support and counseling are absolute necessities for the treatment of anxiety and cannot be overemphasized.
Both benzodiazepine and non-benzodiazepine drugs are used to treat anxiety disorders, with the benzodiazepines the most commonly prescribed anxiolytics in the United States. Before the introduction of these compounds in the 1960s, the major drugs used to treat anxiety were primarily sedatives and hypnotics and included the barbiturates and alcohols. These compounds have potent respiratory depressant effects and led to a high incidence of overdose and death. Although the benzodiazepines are not devoid of side effects, they have a wide margin of safety, with anxiolytic activity achieved at doses that do not induce clinically significant respiratory depression. The non-benzodiazepine anxiolytics include buspirone, β adrenergic receptor antagonists, and antidepressants. Although these drugs are efficacious for several anxiety disorders, their use cannot compare to that of the benzodiazepines.
In addition to their use as anxiolytics, the benzodiazepines are also used for the treatment of insomnia, which often accompanies anxiety and depression. It has been estimated that 30% of all adults in the United States have insomnia, characterized by difficulty both initiating and maintaining sleep. Although the benzodiazepines were the mainstay for the treatment of insomnia for many years, newer agents have been developed including the benzodiazepine receptor agonists eszopiclone, zaleplon, and zolpidem, and the melatonin receptor agonist ramelteon. In addition, several over-the-counter antihistamine preparations are useful for insomnia including hydroxyzine and diphenhydramine (see Chapter 14).
This chapter focuses on the pharmacology of the benzodiazepines and related compounds. The pharmacology of the antidepressants is discussed in Chapter 30 and that of β adrenergic receptor antagonists in Chapter 11.
Therapeutic issues related to both anxiety and insomnia are summarized in the Therapeutic Overview Box.
|
Abbreviations |
|
|
CNS |
Central nervous system |
|
GABA |
γ-Aminobutyric acid |
|
5-HT |
Serotonin |
|
IM |
Intramuscular |
|
IV |
Intravenous |
|
Therapeutic Overview |
|
|
Anxiety Disorders |
Insomnia |
|
Benzodiazepines |
Benzodiazepines |
|
Non-benzodiazepine anxiolytics |
Benzodiazepine receptor agonists |
|
• Buspirone |
Melatonin receptor agonists |
|
• β Adrenergic receptor antagonists |
Antihistamines |
|
Antidepressants |
|
|
• Antidepressants |
|
Mechanisms Of Action
Benzodiazepines
The benzodiazepines exert their effects through allosteric interactions at the γ-aminobutyric acid type A (GABAA) receptor. GABAA receptors are pentameric ligand-gated ion channels, and stimulation of these receptors by GABA leads to the influx of Cl- and a resultant hyperpolarization of the postsynaptic cell (see Chapter 1). This hyperpolarization renders the cell less likely to fire in response to an incoming excitatory stimulus, thus mediating the inhibitory effects of GABA throughout the central nervous system (CNS).
GABAA receptors contain primary agonist binding sites for GABA and multiple allosteric sites that can be occupied by numerous pharmacological compounds, as depicted in Figure 31-1. Benzodiazepines bind to one of these modulatory sites, often referred to as the benzodiazepine binding site or benzodiazepine receptor, whereas compounds such as the barbiturates and the poison picrotoxin bind to other sites on the receptor. When benzodiazepines bind to the benzodiazepine binding site, they induce a conformational change in the receptor, resulting in an increased frequency of chloride ion channel opening upon stimulation of the receptor by GABA. They are referred to as positive allosteric modulators because they increase the effect of the natural agonist but have no effect in the absence of agonist.
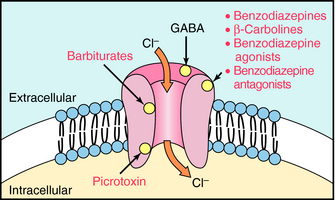
FIGURE 31–1 The GABAA receptor depicting the membrane-associated protein composed of five subunits, the Cl-- channel, and relative location of binding sites for GABA, benzodiazepines, barbiturates, and picrotoxin.
Benzodiazepines are not the only group of compounds that bind to this allosteric site. The β-carbolines such as harmine and harmaline also interact with this site. However, when these compounds bind, they allosterically reduce Cl- conductance by decreasing the affinity of GABA for its binding site. Because the β-carbolines increase CNS excitability and may produce anxiety and precipitate panic attacks, effects opposite to those of the benzodiazepines, they are called inverse agonists (see Chapter 1). The inverse agonists block the effects of the benzodiazepines but have no therapeutic use. However, they are found in nature and are thought to be responsible for the psychedelic properties of some plant species.
Benzodiazepine Antagonist
Flumazenil is a competitive antagonist that binds to the benzodiazepine site on the GABAA receptor. As a competitive antagonist, flumazenil occupies the benzodiazepine site with high affinity but does not have the ability to activate it and does not affect GABA-mediated chloride ion influx. Rather, flumazenil competitively antagonizes the actions of the benzodiazepines and is used therapeutically to treat benzodiazepine overdose. Flumazenil also competitively antagonizes the effects of the inverse agonists and the benzodiazepine receptor agonists, because they also bind at the same allosteric site.
Non-Benzodiazepine Anxiolytics
Buspirone
Buspirone is unrelated chemically to the benzodiazepines or the barbiturates. It is as effective as the benzodiazepines as an anxiolytic but does not have anticonvulsant, muscle relaxant, or sedative effects. Buspirone is a partial agonist at serotonin 5-HT1A receptors, an action that may mediate its anxiolytic effects. It also has a moderate affinity for dopamine D2 receptors, but the relationship between anxiety and dopaminergic activity is unclear. Buspirone has no affinity for either GABA or benzodiazepine binding sites on GABAA receptors.
β Adrenergic Receptor Antagonists
In addition to the benzodiazepines, buspirone, and the antidepressants, β adrenergic receptor antagonists such as propranolol (see Chapter 11) are useful for the treatment of performance anxiety. These compounds suppress the sympathetically-mediated somatic and autonomic symptoms of anxiety.
Benzodiazepine Receptor Agonists
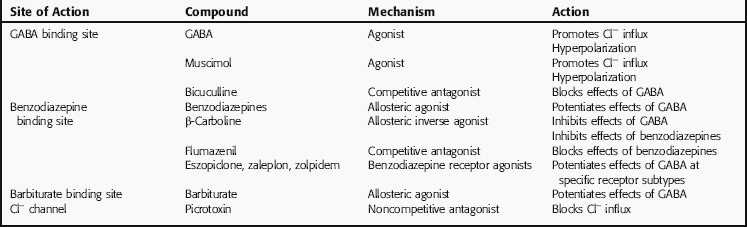
The benzodiazepine receptor agonists eszopiclone, zaleplon, and zolpidem, which are used to treat insomnia, are chemically unrelated to the benzodiazepines but bind to the same site as the benzodiazepines on the GABAA receptor. However, in contrast to the benzodiazepines, which bind to all GABAA receptors irrespective of their subunit composition, the benzodiazepine agonists bind only to a subset of GABAA receptors containing a specific subunit composition. Thus these compounds have a different pharmacological profile than the benzodiazepines (see Relationship of Mechanisms of Action to Clinical Response). A summary of the mechanisms of action of compounds affecting the GABAA receptor is presented in Table 31-1.
TABLE 31–1 Agents Affecting the GABAA Receptor
Melatonin Receptor Agonists
Ramelteon is the first and only melatonin receptor agonist approved for the treatment of insomnia characterized by difficulty falling asleep. Ramelteon, like the hormone melatonin, has high affinity for both melatonin type 1 (MT1) and type 2 (MT2) receptors, both of which are G-protein coupled receptors. MT1 and MT2 are expressed throughout the brain and highly abundant in the suprachiasmatic nucleus of the hypothalamus, an area that is intimately involved in regulating circadian rhythms and sleep and is often referred to as the circadian pacemaker or “master clock.” As a pacemaker, the suprachiasmatic nucleus exhibits an intrinsic rhythm of activity that is inhibited at night as a consequence of the release of melatonin from the pineal gland. Studies suggest that ramelteon, like melatonin, activates melatonin receptors to inhibit the activity of the suprachiasmatic nucleus, leading to the induction of sleep.
Pharmacokinetics
In general, the benzodiazepines are well absorbed after oral administration and reach peak blood and brain concentrations within 1 to 2 hours. Clorazepate is an exception, because it is the only benzodiazepine that is rapidly converted in the stomach to the active product N-desmethyldiazepam. The rate of conversion of clorazepate is inversely proportional to gastric pH.
Whereas the benzodiazepines are typically taken orally, diazepam, chlordiazepoxide, and lorazepam are available for IM and IV injection. Lorazepam is well absorbed after IM injection, but absorption of diazepam and chlordiazepoxide is poor and erratic after this route of injection and should be avoided. When administered IV as an anticonvulsant or for induction of anesthesia, diazepam enters the brain rapidly and is redistributed into peripheral tissues, providing CNS depression for less than 2 hours. In contrast, lorazepam is less lipid soluble and depresses brain function for as long as 8 hours after IV injection.
The duration of action of the benzodiazepines varies considerably, and the formation of active metabolites plays a major role in the effects of these compounds (Table 31-2). The benzodiazepines and their active metabolites are highly bound to plasma proteins, being greatest for diazepam (99%) and lowest for alprazolam (70%). The distribution of diazepam and other benzodiazepines is complicated somewhat by a considerable degree of biliary excretion, which occurs early in their distribution. This enterohepatic recirculation occurs with metabolites and parent compounds and may be important clinically for compounds with a long elimination half-life. The presence of food in the upper bowel delays reabsorption and contributes to the late resurgence of plasma drug levels and activity.
TABLE 31–2 Pharmacokinetic Parameters for Representative Benzodiazepines After Oral Administration
|
Drug |
Onset of Action* |
t1/2† |
|
Alprazolam |
Intermediate |
Intermediate |
|
Chlordiazepoxide |
Intermediate |
Long |
|
Clorazepate |
Rapid |
Long |
|
Diazepam‡ |
Rapid |
Long |
|
Flurazepam |
Rapid |
Long |
|
Halazepam |
Intermediate |
Long |
|
Lorazepam‡ |
Intermediate |
Intermediate |
|
Oxazepam |
Slow |
Short |
|
Prazepam |
Slow |
Long |
|
Temazepam |
Slow |
Intermediate |
|
Triazolam |
Rapid |
Short |
* Rapid = 15-30 min; Intermediate = 30-45 min; Slow = 45-90 min.
† Short, <10 hours; Intermediate, 10-36 hours; Long >48 hours.
‡ Also administered by injection.
The benzodiazepines are metabolized extensively by hepatic microsomal enzymes (Fig. 31-2). The major biotransformation reactions are N-dealkylation and aliphatic hydroxylation, followed by conjugation to inactive glucuronides that are excreted in the urine. The long-acting benzodiazepines clorazepate, diazepam, chlordiazepoxide, prazepam, and halazepam are dealkylated to the active compound N-desmethyldiazepam (nordiazepam). This compound has an elimination half-life of 30 to 200 hours and is responsible for the long duration of action of these compounds. N-desmethyldiazepam is hydroxylated to oxazepam, which forms a glucuronide conjugate. Alprazolam undergoes hydroxylation followed by glucuronidation, and lorazepam is directly glucuronidated.
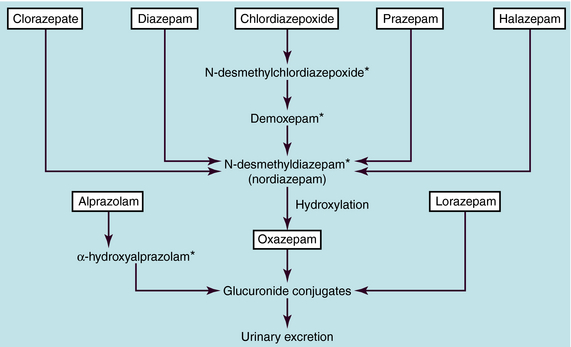
FIGURE 31–2 Major metabolic interrelationships among the benzodiazepines. *Active metabolite.
Flurazepam is a long-acting drug that is converted to desalkylflurazepam, a long-acting active metabolite. Relatively little flurazepam and desalkylflurazepam are excreted unchanged in urine, because they are biotransformed in the liver. Hence their elimination half-lives are long in young adults and even longer in older patients and those with liver disease.
The benzodiazepine receptor antagonist flumazenil is administered IV and has a very short duration of action. It is metabolized by the liver and excreted in the urine with a half-life of approximately 1 hour. Its antagonist activity is manifest within 1 to 2 minutes, a peak effect is seen in 6 to 10 minutes, and its duration of action is approximately 1 hour.
The non-benzodiazepine anxiolytic buspirone is rapidly absorbed and undergoes extensive first-pass metabolism. It is highly bound to plasma proteins and oxidized to an active metabolite. The elimination half-life is approximately 2 to 3 hours, and less than 50% of the drug is excreted in the urine unchanged.
The benzodiazepine receptor agonists, eszopiclone, zolpidem, and zaleplon, have relatively short elimination half-lives of 6, 2.5, and 1 hour, respectively. All these compounds are metabolized extensively, and the inactive metabolites are excreted in the urine. Eszopiclone and zolpidem are metabolized primarily by CYP3A4, whereas this enzyme plays a secondary role in the metabolism of zaleplon, which occurs primarily by aldehyde oxidase. Thus inhibitors of CYP3A4 such as ketoconazole will affect the metabolism of eszopiclone and zolpidem.
The melatonin receptor agonist ramelteon is also rapidly absorbed, undergoes rapid and extensive first-pass metabolism, and is 70% bound to serum albumin. Ramelteon is oxidized primarily by CYP1A2, followed by glucuronidation and urinary excretion, with an elimination half-life of 2 to 5 hours. It should not be used with CYP1A2 inhibitors such as fluvoxamine.
Relationship of Mechanisms of Action to Clinical Response
Benzodiazepines
All the effects of the benzodiazepines are a consequence of their actions in the CNS to enhance GABAergic neurotransmission and thereby cause CNS depression.
Anxiety is managed effectively with the benzodiazepines, particularly alprazolam, lorazepam, and clonazepam. Their intermittent use for acute attacks or limited long-term use (4 to 8 weeks) for recurring symptoms is often beneficial. However, all benzodiazepines should be used cautiously in patients with a history of addiction or more chronic and severe emotional disturbances. Panic attacks respond favorably to alprazolam, which has been shown to possess antidepressant activity similar to that of the tricyclic antidepressants, which are also used for the treatment of panic attacks (see Chapter 30). A debilitating anxiety caused by another illness can be controlled by short-term treatment with anxiolytic drugs while treatment for the primary condition is implemented.
Sedation is the most common effect of the benzodiazepines, and its intensity and duration depend on the dose and concentration of drug in plasma and brain. Although flurazepam, temazepam, and triazolam are used for the treatment of insomnia, they may lead to daytime sedation because of their long duration of action. Oxazepam has a shorter duration of action and would be less likely to cause this problem. The benzodiazepines decrease the latency of sleep onset (time to go to sleep), increase the amount of time spent in stage 2 sleep, and increase total sleep time. However, REM sleep and stage 4 (slow wave) sleep are depressed. If the benzodiazepines are discontinued, a rebound increased REM sleep occurs, characterized by bizarre dreams. Tolerance occurs to the sedative, but not the anxiolytic effect of the benzodiazepines.
Owing to their ability to produce sedation and anterograde amnesia and reduce the anxiety, stress, and tension associated with surgical or diagnostic procedures, benzodiazepines are used both aspreanesthetic medications and for induction and maintenance of anesthesia (see Chapter 35). For procedures that do not require anesthesia, such as endoscopy, cardioversion, cardiac catheterization, specific radiodiagnostic procedures, and reduction of minor fractures, benzodiazepines may be administered orally, IM, or IV.
The benzodiazepines produce skeletal muscle relaxation by inhibiting polysynaptic reflexes. However, most evidence suggests that, with the exception of diazepam, skeletal muscle relaxation occurs only with doses of the benzodiazepines that have significant CNS depressant effects. Diazepam has a direct depressant effect on monosynaptic reflex pathways in the spinal cord and thus produces skeletal muscle relaxation at doses that do not induce sedation. This effect renders diazepam of benefit for relief of skeletal muscle spasms, spasticity, and athetosis.
In addition to these major indications, the benzodiazepines have also been found to be of use for treatment of alcohol withdrawal. Because the benzodiazepines exhibit cross-tolerance with alcohol, have anticonvulsant activity, and do not have major respiratory depressant effects, they have become drugs of choice for treatment of acute alcohol withdrawal symptoms (see Chapter 32). In particular, chlordiazepoxide, lorazepam, and diazepam have now replaced other drugs for this purpose. Choosing between these medications for alcohol withdrawal is often based on metabolic considerations. If a patient has hepatic impairment, lorazepam is often preferred to treat the symptoms of alcohol withdrawal, because it is metabolized in both liver and kidney. If hepatic dysfunction is not an issue, the use of drugs primarily metabolized by the liver (such as chlordiazepoxide and diazepam) would be appropriate.
Although all benzodiazepines act at benzodiazepine binding sites on GABAA receptors, they differ in their pharmacological profiles. For example, some anxiolytic benzodiazepines are non-sedating, whereas other benzodiazepines are used selectively for their sedative properties. Similarly, the incidence of muscle relaxation differs among compounds, and not all benzodiazepines have anticonvulsant activity. Such differences may be attributed to two primary factors, the type of GABAA receptor involved and the nature of the interaction of the benzodiazepine with its binding site.
Current evidence indicates that multiple GABAA receptors exist in the brain. These receptors have different subunit compositions and different anatomical distributions. Studies have shown that receptors containing α2 subunits may mediate anxiolytic effects of the benzodiazepines, whereas other effects (sedation, skeletal muscle relaxation) may be mediated by receptors containing α1 subunits. This may explain why eszopiclone, zaleplon, and zolpidem, which have full agonist activity at receptors containing α1 subunits, are sedating but not anxiolytic.
In addition to receptor subtype selectivity, the divergent pharmacological and behavioral profiles of the benzodiazepines may be explained on the basis of differences in intrinsic efficacy. The benzodiazepines exhibit a broad range of intrinsic efficacies, and studies have shown that partial agonists with anxiolytic and anticonvulsant activities are non-sedating and do not cause muscle relaxation. Thus, as we learn more about benzodiazepine receptor subtypes and the nature of the interactions between these receptors and drugs, therapeutic compounds with selective and specific pharmacological and behavioral profiles may be developed.
Benzodiazepine Antagonist
As mentioned, flumazenil is a competitive antagonist at benzodiazepine receptors and is approved for use to reverse the sedative effects of the benzodiazepines after overdose, anesthesia, or sedation for brief surgical or diagnostic procedures. Flumazenil has also been shown to reverse the sedative effects of the benzodiazepine receptor agonists. Flumazenil does not have any activity of its own and does not antagonize the effects of the opioids, general sedatives, or anesthetic agents. Flumazenil is of great benefit in cases of overdose, and it has been reported that in unconscious adults, flumazenil causes regaining of consciousness sufficient such that gastric lavage, bladder catheterization, electroencephalography, and other procedures could be avoided.
Non-Benzodiazepine Anxiolytics
Buspirone
Buspirone offers an attractive alternative to benzodiazepines for the long-term therapy of less severe and longer lasting forms of anxiety such as generalized anxiety disorder. It produces little sedation and does not produce physical or psychological dependence. This makes buspirone especially useful in patients with a history of substance abuse or dependence. However, the onset of its anxiolytic activity is delayed, making it less suitable for control of an acute anxiety attack. Buspirone is indicated for the management of generalized anxiety disorder and may be more effective than benzodiazepines for chronic states of anxiety in which irritability and hostility are manifest. However, patient compliance is often poor, especially in individuals who have been treated previously with benzodiazepines.
β Adrenergic Receptor Antagonists
In addition to the benzodiazepines, buspirone, and the antidepressants, β adrenergic receptor antagonists such as propranolol (see Chapter 11) are useful for the treatment of performance anxiety or “stage fright.” These compounds are effective in suppressing the somatic and autonomic symptoms of anxiety but do not alter emotional symptoms. Interestingly, the α2 adrenergic receptor agonist, clonidine, has also been reported to have anxiolytic properties.
Benzodiazepine Receptor Agonists
The benzodiazepine receptor agonists eszopiclone, zaleplon, and zolpidem, which are chemically unrelated to the benzodiazepines, produce sedation without anxiolytic, anticonvulsant, or muscle relaxant effects. These compounds have been shown to be highly efficacious for the treatment of transient and chronic insomnia and decrease sleep latency and increase total sleep time without affecting REM sleep. In patients who often need to ambulate in the night, zaleplon is often preferred because of its shorter duration of action; it causes less confusion and less somnolence on awakening. These compounds have been reported to be devoid of abuse potential, because high doses induce nausea and vomiting. However, there is some evidence of addictive effects of zaleplon and zolpidem, but not eszopiclone.
Melatonin Receptor Agonists
The sleep-promoting actions of ramelteon appear to be related to its ability to mimic the endogenous hormone melatonin. Ramelteon has been shown to produce a significant decrease in sleep latency but does not affect sleep maintenance. Ramelteon should be taken within 30 minutes of going to bed and should not be taken with or immediately after a high-fat meal, because its absorption will be delayed. Studies have shown that ramelteon decreases sleep latency by 8 to 16 minutes, is devoid of residual effects the day after administration, and does not produce rebound insomnia.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Benzodiazepines
Clinical problems associated with the use of the benzodiazepines are summarized in the Clinical Problems Box.
General Effects
The adverse reactions most frequently encountered with benzodiazepine use are an extension of their CNS depressant effects and include sedation, lightheadedness, ataxia, and lethargy. The mild sedative actions of these drugs vary quantitatively. For example, lorazepam has a prolonged sedative action compared with the other benzodiazepines, even though it clears the body rapidly. Occasional reactions observed with hypnotic doses of the benzodiazepines include impaired mental and psychomotor function, confusion, euphoria, delayed reaction time, uncoordinated motor function, dysarthria, headache, and xerostomia. Rare reactions may include syncope, hypotension, blurred vision, altered libido, skin rashes, nausea, menstrual irregularities, agranulocytosis, lupus-like syndrome, edema, and constipation.
Anterograde memory disturbances have been observed in patients taking diazepam, chlordiazepoxide, and lorazepam. Thus patients cannot recall information acquired after drug administration. This effect has been attributed to interference with the memory consolidation process and may be beneficial when the benzodiazepines are administered parenterally for presurgical or diagnostic procedures such as endoscopy. In this situation patients should be warned of this effect, especially if they are being treated on an outpatient basis. When administered orally, most benzodiazepines do not cause this effect.
Adverse reactions associated with the IV use of benzodiazepines include pain during injection, thrombophlebitis, hypothermia, restlessness, cardiac arrhythmias, coughing, apnea, vomiting, and a mild anticholinergic effect. Deaths from overdose rarely occur. Patients have taken as much as 50 times the therapeutic doses of benzodiazepines without causing mortality. This particular property of these drugs is another example of how they differ from the potent respiratory depressant sedatives and hypnotics. Unlike the barbiturates, the benzodiazepines have only a mild effect on respiration when given orally, even with toxic doses. However, when they are administered parenterally or are taken in conjunction with other depressants such as alcohol, all benzodiazepines have the potential of causing significant respiratory depression and death.
Drug Withdrawal and Dependence
The benzodiazepines are well known to produce physical dependence, and withdrawal reactions ensue upon abrupt discontinuation. However, the dependence associated with the benzodiazepines is not the same as that observed with alcohol, narcotics, or the barbiturates. Although physical dependence is more likely to occur with high drug doses and long-term treatment, it has also been reported after usual therapeutic regimens. The onset of withdrawal symptoms is related to the elimination half-life and is more rapid in onset and more severe after discontinuation of the shorter acting benzodiazepines such as oxazepam, lorazepam, and alprazolam. With the longer acting benzodiazepines, the onset of withdrawal is much slower because of their longer half-lives and slower disappearance from the plasma. Therefore doses of the benzodiazepines with short half-lives should be decreased more gradually. In addition, alprazolam, estazolam, and triazolam, which have a chemical structure (triazolo ring) that differs from the other benzodiazepines and are referred to as triazolobenzodiazepines, cause more serious withdrawal reactions than the other compounds.
The withdrawal symptoms accompanying abrupt discontinuation from the benzodiazepines are generally autonomic and include tremor, sweating, insomnia, abdominal discomfort, tachycardia, systolic hypertension, muscle twitching, and sensitivity to light and sound. In rare instances severe withdrawal reactions may develop, characterized by convulsions. These reactions are usually manifest in individuals maintained on high doses of the benzodiazepines for prolonged (more than 4 months) periods of time. In addition to these autonomic manifestations, abrupt withdrawal of benzodiazepines can often cause patients to “rebound,” exhibiting symptoms of anxiety and insomnia sometimes worse than before drug treatment was initiated.
The benzodiazepines have been reported to have a high abuse potential. However, evidence suggests that psychological dependence occurs mainly in people with a history of drug abuse; appropriate therapeutic use by persons not predisposed to drug abuse should not lead to abuse of the benzodiazepines.
Drug Interactions
The benzodiazepines are powerful CNS depressants, and additive effects are apparent when they are administered with other CNS depressants. These include ethanol, antihistamines, other sedative/hypnotic agents, antipsychotics, antidepressants, and narcotic analgesics. Because ethanol is readily available and widely used, the CNS depressant interaction between the benzodiazepines and ethanol is common. Individuals may experience episodes of mild to severe ataxia and “drunkenness” that severely retards performance levels. No single compound is considered safer than another in combination with ethanol. Therefore it is imperative that physicians caution their patients not to drink alcoholic beverages while taking these compounds. This is especially important for patients not exposed previously to the benzodiazepines. Individuals who have been drinking alcohol and taking benzodiazepines for long periods of time experience this interaction, but to a milder degree.
Another drug interaction is a consequence of the biotransformation of the benzodiazepines. Because many of the benzodiazepines are metabolized by hepatic microsomal enzymes, therapeutic agents that inhibit CYPs decrease the biotransformation of the long-acting compounds. The histamine H2 receptor antagonist cimetidine and oral contraceptives prolong the elimination half-life of the benzodiazepines by inhibiting their metabolism. Cisapride is a potent inhibitor of CYP3A4 and can cause significant increases in blood levels of many medications, including alprazolam, midazolam, and triazolam. Conversely, compounds that induce this enzyme, such as the barbiturates and carbamazepine, increase their rate of metabolism. Of course, the biotransformation of benzodiazepines that proceed by a route other than hepatic oxidation is unaltered. Although the benzodiazepines are biotransformed via the hepatic microsomal enzymes, they do not significantly induce CYP activity and do not accelerate the metabolism of other agents biotransformed via this system.
Contraindications and Precautions
As with any drug or class of drugs, the benzodiazepines should be avoided in patients with a known hypersensitivity to these agents. Alprazolam, clorazepate, diazepam, halazepam, lorazepam, and prazepam are contraindicated in individuals with acute narrow-angle glaucoma because of their anticholinergic side effects. In addition, because of the considerable lipid solubility of most benzodiazepines, they cross the placenta and are secreted in mother’s milk. It should be noted that the benzodiazepines may be teratogenic and should be avoided in pregnant and nursing women.
Again, because of the hepatic biotransformation of these compounds, special care must be taken when prescribing benzodiazepines for individuals with hepatic dysfunction and for the elderly and debilitated population. These patients generally have a diminished liver-detoxifying capacity and often show cumulative toxicity in response to the usual adult dosage, especially of agents metabolized to active metabolites with long half-lives (diazepam, chlordiazepoxide). The elderly are also more prone to acute depression of attention, alertness, motor dexterity, and sensory acuity as well as memory disturbance and confusion. For this reason doses in the elderly should be started at 25% of the usual adult dose and administered less frequently.
As with other psychoactive medications, precautions should be given with respect to administration of the drug and the amount of the prescription for severely depressed patients or for those in whom there is reason to expect concealed suicidal ideation or plans.
Benzodiazepine Receptor Antagonist
Adverse reactions to flumazenil include nausea, dizziness, headache, blurred vision, increased sweating, and anxiety. In addition, panic attacks have been reported to occur in some patients. Flumazenil can precipitate convulsions in individuals physically dependent on benzodiazepines and in patients maintained on benzodiazepines for seizure disorders. Flumazenil must be used with caution in patients taking benzodiazepines and tricyclic antidepressants, because it may antagonize the anticonvulsant effect of the benzodiazepines and unmask the epileptogenic effect of the tricyclic antidepressant. Cardiac arrhythmias have been reported in some instances.
Buspirone
Adverse reactions of buspirone include dizziness, drowsiness, dry mouth, headaches, nervousness, fatigue, insomnia, weakness, lightheadedness, and muscle spasms. When administered chronically, buspirone causes less tolerance and potential for abuse than the benzodiazepines and does not produce a rebound effect after discontinuation.
Benzodiazepine Receptor Agonists
The most frequent side effects associated with the use of eszopiclone, zolpidem, and zaleplon are headache and dizziness, which appear to be dose-related. In addition, eszopiclone and zaleplon may cause chest pain and anticholinergic effects. Zaleplon may produce nervousness and difficulty concentrating, whereas zolpidem appears devoid of these effects but may lead to confusion and ataxia. There have also been reports of zolpidem inducing delirium, psychotic reactions, and nightmares. The most common side effect of eszopiclone is an unpleasant taste; patients have also reported abnormal dreams and hallucinations.
The abuse liability of zaleplon and zolpidem is less than that of the benzodiazepines when used at the doses recommended. However, when used at higher doses, zolpidem may lead to some physical dependence, and abrupt discontinuation may lead to withdrawal, although less severe than that observed with the benzodiazepines. There is no evidence for abuse liability with eszopiclone.
CLINICAL PROBLEMS
Benzodiazepines
General effects
Sedation, lightheadedness, ataxia, lethargy
Anterograde amnesia
Drug dependence
Addiction liability
Physical dependence with autonomic withdrawal symptoms
Rebound anxiety
Drug interactions
Potentiates CNS depressant effects of alcohol, antihistamines, antipsychotics, antidepressants and opioids
Elimination half-life prolonged by histamine receptor antagonists and CYP3A4 inhibitors
The benzodiazepine agonists, like the benzodiazepines, can produce anterograde amnesia, and as powerful CNS depressants, are additive with other CNS depressants.
Melatonin Receptor Agonists
Ramelteon, unlike the benzodiazepines and benzodiazepine receptor agonists, is not a controlled drug, and there are no reports of rebound insomnia, tolerance, or withdrawal. The most common side effects are somnolence, headache, fatigue, and dizziness. Ramelteon has been reported to be associated with decreased testosterone and increased prolactin levels, although the clinical significance of these alterations is unclear.
New Horizons
There is a need for newer, better tolerated, and more efficacious treatments for anxiety and insomnia, especially drugs without abuse potential. To this end, our understanding of the role of different GABAAreceptor subtypes in the brain is of paramount importance. If each of the pharmacological actions of the benzodiazepines could be ascribed to a specific receptor subtype, then it may be possible to develop compounds with selective actions on these receptors.
Additional approaches to the development of newer compounds depend on our understanding the molecular and cellular events mediating the pathophysiology of stress and stress-related disorders. During the past several years, studies have suggested that the ability to cope with stress involves corticotrophin-releasing factor signaling pathways and that these peptides and their receptors play a major role in generating stress responses. This may represent a new target for development of anxiolytic compounds.
Similarly, as more is learned about the cellular and molecular events regulating specific functional pathways in the brain, research should provide better agents to treat sleep disorders. Sleep-promoting fatty acid amides, neurosteroids, prostaglandins, and peptides are of special interest. New sleep-promoting treatments promise to be more selective in producing natural sleep without disturbing the normal sleep cycle.
TRADE NAMES
(In addition to generic and fixed-combination preparations, the following trade-named materials are some of the important compounds available in the United States.)
Anxiolytics
Benzodiazepines
Alprazolam (Xanax)
Chlordiazepoxide (Librium)
Clonazepam (Klonopin)
Clorazepate (Tranxene)
Diazepam (Valium)
Halazepam (Paxipam)
Lorazepam (Ativan)
Oxazepam (Serax)
Prazepam (Centrax)
Non-benzodiazepine anxiolytics
Buspirone (BuSpar)
Propranolol (Inderal)
Drugs for Insomnia
Benzodiazepines
Estazolam (ProSom)
Flurazepam (Dalmane)
Quazepam (Doral)
Temazepam (Restoril)
Triazolam (Halcion)
Benzodiazepine receptor agonists
Eszopiclone (Lunesta)
Zaleplon (Sonata)
Zolpidem (Ambien)
Melatonin receptor agonist
Ramelteon (Rozerem)
Benzodiazepine receptor antagonist
Flumazenil (Mazicon, Romazicon)
FURTHER READING
Anonymous. Drugs for psychiatric disorders. Treat Guidel Med Lett. 2006;4:35-46.
Anonymous. Treatment of insomnia. Treat Guidel Med Lett. 2006;4:5-10.
SELF-ASSESSMENT QUESTIONS
1. The proposed mechanism of CNS depression by benzodiazepines is:
A. A decreased release of norepinephrine from brain locus coeruleus neurons.
B. A decreased Na+ ion influx at channels located in neuronal postsynaptic membranes.
C. A facilitated GABAA-receptor activity to open chloride ion channels.
D. An antagonist activity at excitatory glutamate receptors.
E. None of the above.
2. Which of the following is least sedative, will NOT potentiate the effects of alcohol, and has no appreciable dependence liability?
A. Chlordiazepoxide
B. Amobarbital
C. Alprazolam
D. Meprobamate
E. Buspirone
3. The mechanism of anxiolytic activity of buspirone is proposed to relate to:
A. A direct action on Cl- channels to enhance hyperpolarizing effects at brain inhibitory synapses.
B. A decrease in muscarinic cholinergic function in the brain “punishment” regions.
C. A partial agonist activity at brain 5-HT1A receptors.
D. An agonist activity at brain adrenergic receptors.
E. An antagonist activity at brain dopaminergic receptors.
4. Diazepam resembles other general CNS depressant drugs in:
A. Promoting psychological dependence.
B. Leading to the development of seizures on sudden withdrawal after long-term treatment with large doses.
C. Demonstrating a cross-dependence pattern to alcohol.
D. All of the above are correct.
E. Both A and C are correct.