PART III
CONGENITAL ANOMALIES AND PEDIATRIC PLASTIC SURGERY
CHAPTER 23 CRANIOSYNOSTOSIS SYNDROMES
SCOTT P. BARTLETT AND CHRISTOPHER A. DERDERIAN
Craniosynostosis, or premature closure of cranial vault and cranial base suture, can involve any suture. Those most commonly observed, in order of decreasing frequency, are sagittal, coronal, metopic, and lambdoidal (Chapter 22). In simple craniosynostosis, one suture is prematurely fused. In multiple-suture synostosis, two or more sutures are prematurely fused. Craniosynostosis can occur as an isolated event resulting in non-syndromic craniosynostosis, or it can occur in conjunction with other anomalies in well-defined patterns that make up clinically recognized syndromes. Syndromic craniosynostosis is most often genetic in nature, and patterns of autosomal dominant, autosomal recessive, and X-linked inheritance have been observed. More than 90 reported syndromes are associated with craniosynostosis, with most involving associated anomalies of the limbs, ears, and cardiovascular system.
The Apert, Crouzon, Pfeiffer, Saethre-Chotzen, and Muenke syndromes represent the more commonly identified craniosynostosis syndromes seen by plastic surgeons. These familial craniosynostosis syndromes share many common features, including midface hypoplasia, cranial base growth abnormalities, abnormal facies, and limb abnormalities. In fact, the craniofacial features are clinically similar among the various syndromes so that the anomalies of the hands may be the differentiating clinical feature between the various syndromes. Although it is clear that synostosis of the cranial sutures is significantly involved in the development of the abnormal craniofacial features in these syndromic children, there probably exists a mesenchymal defect in the cranial base that also contributes to the craniofacial deformity.
The exact etiology of the craniosynostosis in these syndromic children remains unclear. Advances in molecular genetics provide insights into a possible link between mutations identified in fibroblast growth factor receptor (FGFR) genes and several autosomal dominant skeletal disorders. Fibroblast growth factors participate in the regulation of cell proliferation, differentiation, and migration and play a role in controlling normal bone morphogenesis via complex cell-signaling pathways. The transduction of a fibroblast growth factor signal to the cytoplasm is mediated by a group of transmembrane tyrosine kinase receptors known as the FGFRs. Mutations in three of the four known FGFR genes located on chromosomes 8, 10q, and 4p have been identified in the Pfeiffer, Apert, Crouzon, Muenke, and Jackson-Weiss syndromes. Achondroplasia, a skeletal disorder that causes the most common form of short-limb dwarfism, is also linked to a mutation in the FGFR complex. The Pfeiffer syndrome is linked to a mutation in both the FGFR1 and FGFR2 genes, whereas the Crouzon and the Apert syndromes are linked to mutations in the FGFR2 alone. The complexity of this system is highlighted by the observation that two distinct phenotypes of Apert syndrome are caused by missense substitutions in the base sequence for adjacent amino acids in the FGFR2 gene resulting in a higher incidence of cleft palate in one (S252W) and more severe syndactylism in the other (P253R).
CROUZON SYNDROME (ACROCEPHALOSYNDACTYLY TYPE II)
Crouzon syndrome is characterized by premature fusion of calvarial sutures, midface hypoplasia, shallow orbits, ocular proptosis, and FGFR-2 receptor mutation (Figure 23.1). The clinical features were first described by Crouzon, a French neurologist, in 1912. The pattern of inheritance is autosomal dominant. The reported frequency is 1 in 25,000 live births. The variability in expression of the dominant features that make up Crouzon syndrome is widely recognized, and mild deformities in a previously undiagnosed parent of a more severely affected child is a common scenario.
Premature fusion of both coronal sutures, resulting in a brachycephalic head, is the most common calvarial deformity, but scaphocephaly and trigonocephaly, as well as the cloverleaf skull deformity, have been observed, as has “normocephalic” pancraniosynostosis. The craniosynostosis is typically complete by 2 to 3 years of age, but often the sutures are fused at birth. The cranial base sutures are frequently involved, resulting in maxillary and midface hypoplasia. The maxillary hypoplasia is evidenced by a reduced dental arch width and a constricted, high palatal arch. Normal or near-normal mandibular growth leads to a class III malocclusion. The midface hypoplasia is reflected in the shallow orbits with exorbitism, which is a consistent finding and can result in exposure conjunctivitis or keratitis. Exorbitism can be so severe that herniation of the globe through the eyelids may occur, requiring immediate reduction. Acuity problems, strabismus, and hypertelorism have all been reported. A conductive hearing deficit is not uncommon. The defining characteristic of Crouzon syndrome is that no commonly reported limb anomalies are present in this population of syndromic craniosynostosis patients.
Several reports have identified Crouzon syndrome as carrying a higher risk of elevated intracranial pressure (ICP) than other forms of syndromic craniosynostosis, with one study demonstrating a 65% incidence of elevated ICP and the remainder borderline elevated. A retrospective review of suture patency in patients with syndromic craniosynostosis may offer some insight. The review demonstrated that patients with Crouzon syndrome demonstrated earlier closure of the lambdoid and sagittal sutures (median 6 and 21 months, respectively) and a 72% incidence of type I Chiari malformation as compared with patients with Apert syndrome with later suture closure (51 and 60 months, respectively) and a 2% incidence of type I Chiari malformation. We agree with others that these clinically significant characteristics are likely related and should be considered when planning the sequence for surgical treatment of these patients.

FIGURE 23.1. Crouzon syndrome in a young female. Note the midface hypoplasia, shallow orbits, and ocular proptosis. A. Frontal view. B. Profile view.
APERT SYNDROME (ACROCEPHALOSYNDACTYLY TYPE I)
Apert, in 1906, described a syndrome characterized by craniosynostosis, exorbitism, midface hypoplasia, and symmetric syndactyly of both hands and feet. Recently mutations in genes coding for FGFR-2 have been identified in patients with Apert syndrome (Figure 23.2). The incidence is reported to be between 1 in 100,000 and 160,000 live births. Most cases are sporadic, although several cases with autosomal dominant transmission have been reported. The cranial vault deformity in these patients is variable, but most often presents as a short anteroposterior dimension with craniosynostosis involving the coronal sutures resulting in a turribrachycephalic skull, with a large anterior fontanelle. The typical craniofacial appearance includes a flat, elongated forehead with bitemporal widening and occipital flattening. The midface hypoplasia is accompanied by orbital proptosis, downslanting palpebral fissures, and hypertelorism. The nose is downturned at the tip, the bridge is depressed, and the septum is deviated (parrot beak deformity).
The maxillary hypoplasia results in a class III malocclusion with an anterior open bite, and commonly results in significant enough airway compromise to warrant tracheostomy. All patients have a high arched palate and 30% have an associate cleft palate. The hand syndactyly, which is pathognomic for the condition, most often involves fusion of the index, middle, and ring fingers, resulting in mid-digital hand mass; the thumb and little fingers may also be joined to the mid-digital mass (Figure 23.3). When the thumb is free, it is broad and deviates radially due to an accompanying delta phalanx. In the feet, the syndactyly also usually involves the second, third, and fourth toes. These hand anomalies are so severe and functionally debilitating that referral to a hand surgeon with special expertise in this area is essential. An extensive review of central nervous system problems in patients with Apert syndrome shows an increased incidence of delayed mental development, but many of these patients develop normal intelligence. Acne vulgaris is another characteristic feature seen during adolescence in over 70% of patients.
Perhaps our best understanding of the natural history of raised ICP in Apert syndrome comes from the Great Ormond Street Hospital data on the expectant management of their patients. Their protocol is to offer cranial vault expansion only in the setting of confirmed elevation of ICP. Raised ICP developed in 83% (20/24) of patients, 50% in the first year of life, with the average age of onset at 18 months (range 1 month to 4 years 5 months). Thirty-five percent of those who were treated successfully for their first episode of elevated ICP went on to develop a second episode on average 3 years 4 months later.

FIGURE 23.2. Apert syndrome in a 2-year-old female. Note the severe midface hypoplasia, elongated forehead with temporal widening, brachycephaly, and beaked nose.

FIGURE 23.3. A and B demonstrate the characteristic hand and foot syndactyly, which are pathognomic for Apert syndrome. This complex syndactyly most often involves fusion of the second, third, and fourth fingers, resulting in mid-digital hand mass, but the first and fifth fingers may also be joined to the mid-digital mass. In the feet, the syndactyly also usually involves the second, third, and fourth toes.
PFEIFFER SYNDROME (ACROCEPHALOSYNDACTYLY TYPE V)
This syndrome was described by Pfeiffer in 1964 and consists of craniosynostosis, broad thumbs, broad great toes, and, occasionally, a partial syndactyly involving the second and third digits.Pfeiffer syndrome is linked to mutations in both FGFR-1 and FGFR-2, which confer less and more severe craniofacial dysmorphism, respectively. Symptoms vary, ranging from very mild to severe. The mode of inheritance is autosomal dominant. The craniofacial features are similar to those of Apert or Crouzon syndrome. The skull is turribrachycephalic secondary to the coronal and occasional sagittal synostosis (Figure 23.4). Maxillary hypoplasia with resulting midface deficiency leads to shallow orbits and exorbitism. Hypertelorism and downslanting palpebral fissures are also common. The nose is often downturned with a low nasal bridge. Intelligence is reported to be normal in the more common form of Pfeiffer syndrome. Broad thumbs and great toes are the hallmark of the syndrome, but the findings are frequently subtle. The partial syndactyly of the hands usually involves the index and middle fingers. A partial syndactyly of toes 2, 3, and 4 has also been noted. Cohen proposed a classification system that clusters patients into three types based upon their clinical findings and severity. Type I represents the “classic Pfeiffer” syndrome (features described above) which is milder than type II with a kleeblattschädel (cloverleaf) skull, and type III Pfeiffer syndrome is the most severely affected.

FIGURE 23.4. Pfeiffer syndrome in an infant female. A. Note the severe midface hypoplasia, exorbitism, mild hypertelorism, and turribrachycephalic skull. B. The profile view clearly demonstrates the abnormal brow-to-cornea relationships, concavity of the midface, and short nose.
A recent review of 28 patients treated at a single institution reported that the Cohen subtypes distribution was type 1 61%, type II 25%, and type III 14%(Fearon). These patients underwent an average of 2.5 cranial vault procedures, 1.6 neurosurgical procedures, and 3.5 other operations. In addition to the challenging reconstructive needs of the patient with Pfeiffer syndrome, this study highlighted several functional considerations that should be aggressively treated or monitored in these patients including value in early placement of permanent tarsorrhaphies and supplementing these with temporary tarsorrhaphies at the time of cranial vault procedures, and high incidences of aural atresia (54%), conductive hearing loss (86%), need for tracheostomy (61%), hydrocephalus (68%), and Chiari malformations (82%).
SAETHRE-CHOTZEN SYNDROME (ACROCEPHALOSYNDACTYLY TYPE III)
This syndrome was first described by Saethre in 1931 and by Chotzen in 1932. The predominant features include a brachycephalic skull, a low-set frontal hairline, prominent crus helicis extending through the conchal bowl, facial asymmetry, and ptosis of the eyelids (Figure 23.5). The mode of inheritance is autosomal dominant, with wide variability in expression. The diagnosis is confirmed by identification of a mutation in the TWIST-1 gene on chromosome 7p21, which is believed to result in a dysregulation between bone deposition and maintenance of suture patency.
The craniofacial features include unicoronal or bicoronal synostosis, which is often asymmetric giving a plagiocephalic appearance and contributes to facial asymmetry. The low-set hairline is also a constant feature of this syndrome. The facial asymmetry is often accompanied by deviation of the nasal septum and maxillary hypoplasia with a narrow palate. Intelligence is usually normal. A partial syndactyly involving the index and long fingers is often observed, and short stature is also a frequent finding.

FIGURE 23.5. Saethre-Chotzen syndrome. Note the brachycephalic and turricephalic skull, low-set frontal hairline, and ptosis of the eyelids (A–D). In this case, the patient had multiple-suture craniosynostosis necessitating a strip craniectomy in early infancy. This was followed by fronto-orbital advancement at 10 months to improve brow position and frontal bone contour (E, F), followed by posterior cranial vault distraction at age 24 months to remove occipital flatness and the increased height of the posterior skull while further expanding the intracranial volume (G, H). With a decreased posterior vertical height, the increased anterior vertical height and recurrent brow retrusion are addressed during a second FOA (I–L).
Patients with Saethre-Chotzen have a high incidence of need for reoperation after cranial vault expansion, ranging from 42% to 65%. Several reports have demonstrated high reoperation rates for poor growth after fronto-orbital advancement (FOA); however, a recent report demonstrated that this patient population also carries a greater than 40% risk of developing elevated ICP after the initial cranial vault expansion. Clearly, such a high-risk population warrants strict monitoring and their families should be made aware that the majority of these patients require more than one cranial vault procedure in the course of their treatment.
MUENKE SYNDROME
Unlike other eponymous craniosynostosis syndromes, Muenke syndrome derives its name from the first report of the genetic mutation rather than the phenotype. The mutation is a pro250Arg mutation in FGFR-3 on chromosome 4p, which has an incidence of 1 in 10,000 and demonstrates an autosomal dominant inheritance pattern with variable expressivity. It is estimated that Muenke syndrome may be present in 10% of unicoronal or bicoronal synostosis cases that were previously believed to be non-syndromic in origin. The most consistent features include craniosynostosis of the coronal sutures, hearing loss, developmental delay, and thimble-like middle phalanges. Midface hypoplasia is not a common finding. Muenke syndrome exhibits significant variability in the presentation of craniosynostosis between genders, where 88% of females and 76% of males with the mutation have craniosynostosis. While bicoronal synostosis is the most common presentation for both sexes, males demonstrate a much higher incidence of unicoronal craniosynostosis (37% bicoronal vs. 29% unicoronal) than females (58% bicoronal vs. 20% unicoronal). The pattern of sensorineural hearing loss found in these patients is characteristically a bilateral, symmetric, low- to mid-frequency pattern.
The clinical relevance of Muenke syndrome lies in the course of these patients after their initial surgical treatment. In a large retrospective review for patients with coronal craniosynostosis, the reoperation rate for elevated ICP in Muenke syndrome was five times more common than in those without the mutation. Others have also found rates of reoperation to be much higher and aesthetic outcomes to be poorer in Muenke syndrome. In a patient population that is already at an increased risk for developmental delays, and lack significant extracranial signs of their genetic diagnosis, a high level of suspicion and low threshold for genetic testing must be had when evaluating patients with unicoronal or bicoronal synostosis, particularly those with a family history.
FUNCTIONAL ASPECTS
To fully appreciate the surgical treatment of children with these craniosynostosis syndromes, it is necessary to understand the craniofacial growth process and how it relates to certain functional aspects of development. Normal craniofacial growth is directed by two general processes: displacement and bone remodeling. During the first year of life, the brain triples in size and continues to grow rapidly until about 6 or 7 years of age. The growth of the brain causes displacement of the overlying frontal, parietal, and occipital bones in the presence of open functioning sutures, and this stimulates bone growth and remodeling in the skull and cranial fossa. The growth and maturation of the face follows a craniocaudal gradient, progressing from late childhood to adolescence, with maturation of the upper face followed by maturation of the midface and finally the mandible. The functional aspects of development, which are directly or indirectly influenced by abnormal craniofacial growth, are examined individually below.
Intracranial Pressure
The rapid growth of the brain in infancy demands rapid accommodation of the cranial vault to maintain normal ICP. In the patient with craniosynostosis, there can be restricted growth of the cranial vault, resulting in a disparity between brain size and intracranial volume, which leads to increased ICP.
Increased ICP can be recognized clinically by the finding of papilledema on funduscopic examination and, in later stages, “thumb printing” or the beaten cooper appearance on plain radiographs from pressure of the gyri on the inner table of the calvarium. However, papilledema alone is not a reliable marker for the potential for visual dysfunction nor increased ICP. Intraparenchymal monitoring remains the gold standard in ICP monitoring. The placement of pressure-sensitive filaments through a burr hole in the calvarium allows for continuous monitoring of ICP during normal activities over a 24-hour period or longer; however, it carries the risks of bleeding and infection and requires admission to the hospital.
Unfortunately, there does not yet exist an equally reliable noninvasive indicator of increased ICP. In a study that evaluated ICP by an epidural sensor in 358 children with various types of craniosynostosis, it was found that children with multiple-suture synostosis had higher rates of increased ICP (26% to 54%), and in the syndromic population increased ICP was noted in 66% of the patients with Crouzon syndrome and in 43% of those with Apert syndrome. Although it has been documented by three-dimensional computerized tomography (CT) studies that increases in intracranial and ventricular volume occur following cranial vault reshaping, and that ICP pressure decreases, we cannot yet accurately determine by means of CT scan alone which group of patients with craniosynostosis will develop increased ICP. Several new modalities have potential for noninvasive monitoring of ICP.
The first is transorbital ultrasound measurement of optic nerve sheath diameter. Elevations in ICP affect the space around the optic nerve causing an increase in optic nerve diameter, and elevation above age-controlled diameters can reliably predict elevated ICP. Another promising modality is visual evoked potentials (VEPs). This technique measures the latency time of flash VEP–induced N2 wave response. Prolonged response time is predictive of elevated ICP. These techniques carry no inherent risks but limit monitoring to short observation periods. This may be misleading, because transient changes in ICP can occur based upon the patients’ activity, position, or time of day. Therefore, confirmation with invasive monitoring is still required to correlate these findings. However, the combination of several signs and symptoms is frequently enough, when combined with the dysmorphic features, to warrant surgical intervention for both functional- and appearance-related purposes.
Visual Changes
Craniosynostosis can result in abnormal growth of the skull and, in the syndromic population of patients, this is often accompanied by midface hypoplasia. Underdeveloped shallow orbits or abnormally shaped orbits can cause the eyes and periorbital structures to be displaced from their normal position; this is termed exorbitism. Exorbitism can result in corneal exposure and the development of keratitis, pain, infection, corneal scarring, and, at worst, ulceration and blindness. Occasionally, the degree of exorbitism is so great that immediate surgical intervention is required to protect the globe. Ocular motility problems frequently arise secondary to the abnormal size and shape of the orbits. Strabismus with exotropia is a common finding. Abnormal development and position of ocular muscles have also been frequently reported in children with Crouzon or Apert syndrome.
Increased ICP leading to papilledema and optic atrophy can result in blindness. Whether the optic atrophy is secondary to increased ICP or is secondary to damage to the nerve from compression or a compromised vascular supply is not entirely clear. As in monitoring for increased ICP, transient pattern reversal VEP has emerged as a screening tool for possible visual dysfunction in children with syndromic craniosynostosis.
Hydrocephalus
Although the incidence of hydrocephalus and craniosynostosis is rare, it appears that among children with craniosynostosis syndromes, the incidence of hydrocephalus is significantly higher, with reports ranging from 4% to 10%. There is clearly a higher incidence of hydrocephalus among children with Apert syndrome. The etiology of hydrocephalus remains unclear, but it has been postulated that it is caused by increased venous pressure in the sagittal sinus secondary to obstruction of the venous outflow caused by the craniosynostosis. Both communicating and noncommunicating forms of hydrocephalus have been identified, but the communicating form is more common. Hydrocephalus can present without either marked head enlargement (which may be difficult to detect in the patient with syndromic craniosynostosis) or signs of increased ICP. Preoperative CT scanning or ultrasonography helps define the population at risk. At the earliest sign of progressive ventricular enlargement, a shunting procedure should be performed to prevent cerebral injury.
SURGICAL MANAGEMENT
The surgical treatment of patients with craniosynostosis syndromes dates from the late 19th century, when the first techniques were aimed at correcting only the functional aspects of the deformity. The earliest techniques, linear craniectomy and fragmentation of the cranial vault, still are useful in some of the more severe deformities to provide temporary brain and eye protection until a more definitive craniofacial procedure can be undertaken. Simple craniectomy or morcellation performed in infancy, unfortunately, is accompanied by a high rate of reossification and will give only modest results when mobilization of the orbits and midface is not performed concurrently. Additionally, the reossified bone is of poor quality, making definitive correction more difficult. In 1967, Tessier first published his results following correction of the recessed forehead and supraorbital regions using an intracranial approach that allowed accurate osteotomy, mobilization, and repositioning in adults. The current surgical treatment approach is for children with syndromic craniosynostosis and accompanying midface deficiency and involves an initial posterior cranial vault distraction or FOA and cranial vault remodeling, a midface advancement procedure with or without distraction (Le Fort III or monobloc), and secondary orthognathic surgery to correct any dentofacial deformities (Le Fort I, mandibular osteotomies).
Surgical intervention for the correction of craniofacial deformities in patients with syndromic craniosynostosis can be divided into those procedures that are performed early in life (4 to 12 months) for suture release, cranial vault decompression, and upper orbital reshaping/advancement and those that are performed at a later age (4 to 12 years) for midface deformities and jaw surgery (14 to 18 years). The exact timing and sequence for each of the aforementioned surgical procedures is dependent on both the functional and the psychological needs of the patient (Table 23.1). The area of largest controversy centers around the timing of midface osteotomies. Two approaches are currently practiced: (a) waiting until all midface and lower face growth is complete before performing a definitive osteotomy and advancement, or (b) performing a midface advancement in childhood with the realization that a second advancement will be necessary when mandibular growth is complete. Because midface advancement is usually performed using distraction techniques, the complications of blood loss and infection have been dramatically reduced, making the procedure more common in childhood.

Fronto-orbital Advancement
The surgical goals of FOA are threefold: (a) to release the synostosed suture and decompress the cranial vault, (b) to reshape the cranial vault and advance the frontal bone, and (c) to advance the retruded supraorbital bar, providing improved globe protection and an improved aesthetic appearance. The procedure is performed through a coronal incision. With the assistance of a neurosurgical team, a frontal craniotomy is performed to release the involved sutures and elevate the frontal bone. In certain instances, the child may have undergone a prior frontal craniotomy to release the coronal sutures when elevated ICP was suspected. Reossification usually has occurred by 1 year of age. Once the frontal bone is removed, the brain is gently retracted, exposing the underlying retruded supraorbital bar, which is advanced, bolstered forward with a cranial bone graft, and secured with resorbable plates or sutures (Figure 23.6). Cranial vault remodeling technique is dependent on the preoperative head shape. For severe turricephaly, a total cranial vault reshaping is performed (often done in stages); this procedure allows for a significant reduction in the vertical height of the skull. For the child with mild turricephaly, only the anterior two-thirds of the vault is remodeled. The supraorbital bar and forehead are advanced into an overcorrected position to allow room for further brain growth. In patients aged 12 months or younger, the majority of the cranial gaps created by FOA will ossify spontaneously. The ability to close large calvarial defects changes between 9 and 11 months of age, resulting in an increasingly lower probability of defect closure. Overall, roughly 20% of all patients undergoing FOA will have persistent defects. We have therefore adopted the practice of primary grafting of these defects with particulate bone harvested from the endocortex of the cranial bone flaps as described by Greene et al. (Chapter 22).
Posterior Vault Expansion with Distraction Osteogenesis
Expansion of the posterior cranial vault provides a significantly greater gain in intracranial volume than FOA; however, expansion has historically been limited by soft tissue closure. With the advent of distraction osteogenesis (Chapter 24), the concomitant expansion of the soft tissue envelope in addition to the posterior cranial vault allows for significantly greater volumetric expansion and relief of ICP. This procedure is particularly beneficial to patients with raised ICP who are too young for FOA or for those in whom greater functional and aesthetic gains would be made from a posterior expansion rather than a second FOA. Posterior vault expansion has also been noted to have a positive affect on the appearance of the anterior vault. While definitive remodeling procedures of the anterior vault and supraorbital bar are still necessary, the expansion of the posterior vault provides protection to the rapidly growing brain and allows for a single procedure to be performed for fronto-orbital reconstruction. The procedure, performed through a standard coronal incision, consists of a posterior craniotomy with limited dural dissection, barrel staving at the base of the occiput to limit step-off deformity, application of two collinear 1.5 mm mandibular distraction devices, and closure. Activation is started at 3 to 7 days with advancement at 1 mm/d. Advancements are typically between 20 and 30 mm, followed by a 6- to 8-week consolidation period. A second, limited procedure is required for distraction device removal.
Following these cranial vault remodeling and FOA procedures, the child is seen on a 6- to 12-month basis by the craniofacial team. Continued growth of the cranial vault and midface is monitored closely by means of three-dimensional CT scans, as well as clinical observation.

FIGURE 23.6. A. The typical lines of craniotomy for removal of the frontal bone flaps and lines of osteotomy for the frontal bandeau. The frontal bone flaps are carried posteriorly, behind the position of the coronal sutures and anteriorly to leave sufficient height to the bandeau (≈2 cm). B. Exposure of the temporal fossa and anterior cranial base is provided by the neurosurgeon. Bandeau cuts are carried inferiorly from the coronal craniotomy to the level of the zygomaticofrontal (ZF) suture and anteriorly, parallel to the craniotomy cuts, through the ZF suture. Using malleable retractors to protect the temporal and frontal lobes and orbital contents, osteotomies are made through the orbital roofs and transversely just above the nasofrontal junction.
Although FOA and posterior vault distraction provide excellent decompression of the craniosynostosis and improvement in the shape of the cranial vault in the early postoperative period, continued growth restriction in both the cranial vault and the midface region often produces poor long-term aesthetic results in these syndromic patients. If signs of increased ICP, severe exorbitism, or an abnormally shaped cranial vault recur, a second and, occasionally, a third cranial vault remodeling procedure are indicated (Figure 23.7).
Surgical Correction of the Midface Deformity
The first attempt to correct the midface deformity in a syndromic craniosynostosis patient was a Le Fort III procedure performed by Sir Harold Gillies. He later abandoned the operation because of its morbidity, but the procedure was later resurrected and popularized by Paul Tessier (Figure 23.8). The Le Fort III can be performed alone or, if all permanent teeth have erupted, in conjunction with a Le Fort I advancement. The monobloc frontofacial advancement procedure, which involves the advancement of the Le Fort III fragment in coordination with the frontal bar, was developed by Ortiz-Monasterio (Figure 23.9). The monobloc procedure, while offering the advantage of simultaneously correcting the supraorbital and midface deformity, is associated with greater blood loss and a higher infection rate, which is most likely a result of the direct communication between the cranial and nasal cavities. Patients with ventricular shunts have an exceedingly high rate of infection because the brain cannot expand into the newly created dead space. This increased risk makes the traditional monobloc procedure in the neonatal period contraindicated. However, Polley has demonstrated that a monobloc distraction may be performed early in select patients with significant upper airway obstruction and craniosynostosis.
The exact timing of midface correction remains a controversy among craniofacial surgeons. Some craniofacial centers advocate early surgical correction between the ages of 4 and 7 years; others prefer to wait until skeletal maturity is reached at around puberty, unless airway obstruction or severe exorbitism dictates immediate early surgery. The advocates of delayed surgical correction cite evidence of a high incidence of recurrent class III malocclusion in patients who undergo surgery earlier (4 to 9 years), often requiring a secondary Le Fort III procedure in the teenage years. The authors believe that early correction of the midface deformity affords the patient an overall aesthetic improvement that will have a significant positive psychological effect and improve self-esteem in these children, and in our experience patients accept a secondary Le Fort III or monobloc procedure as a standard step in their treatment.
Regardless of the timing, distraction has evolved as the treatment modality of choice for the extensive midface advancement these patients require. The efficacy and improved safety profile of both Le Fort III and monobloc distraction over single staged osteotomy advancement procedures have been well documented. Distraction allows for expansion of the soft tissue envelope with the bony advancement, which aids in preservation of the barrier between the nasopharynx and anterior vault and decreased tendency to relapse from soft tissue contraction. A modified Le Fort III osteotomy is performed (Figures 23.8 and 23.9). Distraction is initiated at 5 to 7 days post-op (principles of distraction discussed in Chapter 24). The surgical technique and distraction protocol is dictated by the patient’s bony deformity. In addition to Le Fort III osteotomy, the techniques for midface improvement may include facial bipartition to correct the hypertelorism, downslanting palpebral fissures, and midface concavity that are characteristic of Apert and Pfeiffer syndromes.
In our institution, the age of midface distraction is typically 5 years. While the goals of midface distraction in this age group are tailored to the patient’s deformity, the universal goals are maximal projection of the zygoma and restoration of orbital volume. The result is typically a transition from a severe Angle Class III to Class II occlusion. The final occlusion is addressed with the definitive orthognathic procedure once the patient reaches skeletal maturity. Advantages of distraction include (a) less blood loss and shorter operative time at the initial procedure; (b) greater advancement (up to 20 mm or more) as compared with standard advancement techniques (6 to 10 mm maximum); (c) less risk of infection with the monobloc procedures; and (d) less relapse. Disadvantages include the (a) prolonged time (several months) needed for distraction and consolidation; (b) need for a second procedure to remove buried devices; and (c) need for wearing an external halo device for a prolonged period. Overall, distraction osteogenesis has improved the results obtainable for midface advancement while minimizing the complications. Figure 23.10 is an example of a patient who underwent a monobloc advancement utilizing distraction osteogenesis.
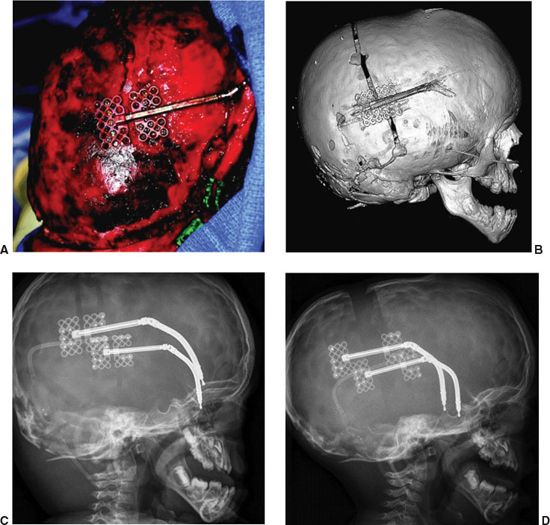
FIGURE 23.7. Posterior vault distraction. In addition to volume expansion of the cranial vault, a second goal of posterior vault distraction is to improve the typically flat occiput of the turricephalic skull. This is accomplished with osteotomy design and vector selection that allows posteroinferior movement of the distracted bone flap as seen in (A) and (B). The dura remains attached to the bone flap and barrel staves are made inferiorly to allow for a smooth transition of the post-distraction bone flap and inferior occiput. To prevent displacement of the device, a minimum of five screws are used per footplate of a 1.5 mm mandibular distraction device with a 30 to 35 mm barrel. The lateral X-rays in the pre- and post-activation states are seen in (C) and (D).

FIGURE 23.8. A. Osteotomies for Le Fort III distraction. Note that the lateral orbital rim osteotomy is in a much lower position than in the traditional Le Fort III procedure. B. This figure demonstrates the final position of the midface segment at the end of the activation phase. With internal devices, it is critical to have collinear positioning of the devices with the correct vector as these devices afford no ability to adjust the vector of distraction during activation.

FIGURE 23.9. A. Osteotomies for monobloc distraction with an external distraction device in place. B. This postoperative view shows the overcorrected position of the midface and depicts the location of generate deposition in the area of the pterygoid plates posterior to the advanced midface segment. While as close an approximation to the desired vector is used in device application, the external device affords significant ability to adjust the vector of distraction in multiple planes. This flexibility makes the external device superior to internal devices in our opinion.
Orthognathic Surgery
The abnormal patterns of facial growth in children with craniosynostosis syndromes often result in significant dentofacial deformities. The class III malocclusion, secondary to midface retrusion, is the most commonly seen deformity and often develops despite appropriate midface surgical treatment. The team approach to the management of these jaw abnormalities involves an orthodontist, a dentist, and a craniofacial surgeon. Following the completion of growth of both the maxilla and the mandible and any needed presurgical orthodontic therapy, surgical correction involving at least a Le Fort I osteotomy with a sliding genioplasty is usually indicated. These surgical procedures are usually performed between the ages of 14 and 18 years, when the facial skeleton is mature.

FIGURE 23.10. Fronto-orbital and midface advancement at age 6 years using monobloc distraction in a child with Crouzon syndrome. A. Preoperative and postoperative frontal view and 3D CT reconstructions. B. Preoperative and postoperative profile view and three-dimensional computerized tomography (CT) reconstructions. Note the desired, overcorrected position of the midface and orbit-producing mild enophthalmos and a significant Angle class II relationship.
Final Facial Contouring
At the completion of facial growth and all major osteotomies, contour irregularities of the facial skeleton may still remain. Final contouring procedures are often performed at this time. They include smoothing irregularities, adding bone grafts or bone substitutes to different areas (e.g., calcium carbonate cements), and resuspending soft tissues such as the midface or lateral canthi.
CONCLUSION
In the past, children with craniosynostosis syndromes were stigmatized as being mentally challenged because of their craniofacial features when, in fact, they were often of normal intelligence. The advent of craniofacial surgery techniques, although far from perfect, offers these children a chance of obtaining a more normal facial appearance and the opportunity to grow, develop, and integrate socially with their peers. The application of newer operative techniques to craniofacial surgery, including endoscopic surgery and distraction osteogenesis, is expected to offer improved results with fewer complications. Distraction osteogenesis has yielded promising results in the cranium and midface. The real future of children with craniosynostosis syndromes, however, lies in the hands of the molecular geneticists. The advances in this field have allowed for the identification of the gene and associated mutation for several craniosynostosis syndromes. Ultimately, the ability to genetically screen for these DNA mutations will allow for appropriate family counseling and perhaps, in the future, gene therapy for the correction of the mutation.
Suggested Readings
1. Arnaud E, Marchac D, Renier D. Reduction of morbidity of the frontofacial monobloc advancement in children by the use of internal distraction. Plast Reconstr Surg. September 2007;120(4):1009-1026.
2. Bartlett SP, Losee JE, Baker SB. Reconstruction: craniofacial syndromes. In: Mathes SJ, ed. Plastic Surgery. Vol IV. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2006:495-520.
3. Bradley JP, Gabbay JS, Taub PJ, et al. Monobloc advancement by distraction osteogenesis decreases morbidity and relapse. Plast Reconstr Surg. December 2006;118(7):1585-1597.
4. Cohen MM Jr, ed. Craniosynostosis: Diagnosis, Evaluation, and Management. New York, NY: Raven Press; 1986.
5. Fearon JA. Halo distraction of the Le Fort III in syndromic craniosynostosis: a long-term assessment. Plast Reconstr Surg. May 2005;115(6):1524-1536.
6. Fearon JA, Rhodes J. Pfeiffer syndrome: a treatment evaluation. Plast Reconstr Surg. May 2009;123(5):1560-1569.
7. Flores RL, Shetye PR, Zeitler D, et al. Airway changes following Le Fort III distraction osteogenesis for syndromic craniosynostosis: a clinical and cephalometric study. Plast Reconstr Surg. August 2009;124(2):590-601.
8. Foo R, Guo Y, McDonald-McGinn DM, Zackai EH, Whitaker LA, Bartlett SP. The natural history of patients treated for TWIST1-confirmed Saethre-Chotzen syndrome. Plast Reconstr Surg. December 2009;124(6):2085-2095.
9. Gosain AK, Santoro TD, Havlik RJ, et al. Midface distraction following Le Fort III and monobloc osteotomies: problems and solutions. Plast Reconstr Surg. 2002;109:1797-1808.
10. Greene AK, Mulliken JB, Proctor MR, Rogers GF. Primary grafting with autologous cranial particulate bone prevents osseous defects following fronto-orbital advancement. Plast Reconstr Surg. November 2007;120(6):1603-1611.
11. Honnebier MB, Cabiling DS, Hetlinger M, McDonald-McGinn DM, Zackai EH, Bartlett SP. The natural history of patients treated for FGFR3-associated (Muenke-type) craniosynostosis. Plast Reconstr Surg. March 2008;121(3):919-931.
12. Liasis A, Nischal KK, Walters B, et al. Monitoring visual function in children with syndromic craniosynostosis: a comparison of 3 methods. Arch Ophthalmol. August 2006;124(8):1119-1126.
13. Paige KT, Vega SJ, Kelly CP, et al. Age-dependent closure of bony defects after frontal orbital advancement. Plast Reconstr Surg. September 2006;118(4):977-984.
14. Shetye PR, Boutros S, Grayson BH, McCarthy JG. Midterm follow-up of midface distraction for syndromic craniosynostosis: a clinical and cephalometric study. Plast Reconstr Surg. November 2007;120(6):1621-1632.
15. Slaney SF, Oldridge M, Hurst JA, et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. May 1996;58(5):923-932.
16. Steinbacher DM, Skirpan J, Puchała J, Bartlett SP. Expansion of the posterior cranial vault using distraction osteogenesis. Plast Reconstr Surg. February 2011;127(2):792-801.
17. Tessier P. The definitive plastic surgical treatment of the severe facial deformities of craniofacial dysotosis: Crouzon’s and Apert’s disease. Plast Reconstr Surg. 1971;48:419.
18. Thompson DN, Harkness W, Jones B, Gonsalez S, Andar U, Hayward R. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst. May 1995;11(5):269-275.
19. Whitaker LA, Bartlett SP, Shut L, et al. Craniosynostosis: an analysis of the timing, treatment, and complications in 164 consecutive patients. Plast Reconstr Surg. 1985;80:195.
20. White N, Evans M, Dover MS, Noons P, Solanki G, Nishikawa H. Posterior calvarial vault expansion using distraction osteogenesis. Childs Nerv Syst. February 2009;25(2):231-236.